
Novel molecular players of X chromosome inactivation: new technologies and new insights


Abstract
The dosage compensation in placental mammals is achieved by silencing of one copy of the X chromosomes in a female cell by a process called X chromosome inactivation (XCI). XCI ensures equal gene dosage for X-linked genes between the two genders. Although the choice of X chromosome to be silenced is random, once the silencing of the X chromosome has been established, this process is highly regulated and maintained throughout subsequent cell divisions. A long non-coding RNA, Xist, and its interacting proteins execute this multistep process, but several of these regulatory proteins remain unidentified. Recent technological advances based on the genetic and proteomics screening have identified several new regulatory factors as well as dissected the molecular details of XCI regulation. Moreover, identification of regulators of XCI offers an opportunity to explore reactivation of the inactive X chromosome (Xi) as a potential therapeutic strategy to treat X-linked diseases, like Rett syndrome. Here, we summarize recent reports that identified new regulatory proteins and RNA species playing a crucial role in Xist localization and spreading, recruitment of silencing machinery to the Xi, Xist interaction with chromatin, and structural organization of the Xi in the nuclei.
Keywords
Introduction
X chromosome inactivation (XCI) is an epigenetic silencing phenomenon that achieves dosage compensation in mammals. First described by Lyon[1] in 1961, XCI is established during early embryonic development, and the fixed inactivation is then inherited through subsequent divisions in somatic cells. Ultimately, XCI balances gene dosage by ensuring that only one X chromosome is transcriptionally active per diploid cell. However, the molecular details of the well-coordinated silencing of the X chromosome are not completely understood.
The mammalian XCI is a multistep process and requires multiple regulatory factors for its successful execution. However, gene dosage compensation is not unique to mammals, but also occurs in invertebrate species, such as Drosophila melanogaster and Caenorhabditis elegans. While D. melanogaster doubles transcription from the X chromosome in XY males, the X-linked gene expression is halved in XX C. elegans females to equalize gene expression amongst genders[2,3]. Even though the mechanistic strategies are substantially different among species, considerable parallels are observed in the mechanisms by which dosage compensation is achieved. Based on the investigation in different models, XCI has three well-demarcated stages: initiation, establishment and maintenance[4]. Lack of molecular details has been a major roadblock in understanding how this complex process of XCI is regulated. The key players required for maintaining stringent control of the transcriptional state of only one X chromosome in female cells remain mostly unidentified. Importantly, defining the mechanism of XCI will also shed light on some of the unaddressed questions in the field. For instance, one of the major challenges is how the compensation machinery discriminates between two essentially identical X chromosomes. What also remains unanswered is the mechanism that precisely controls the counting of the X chromosomes and the random choice of only one X chromosome for inactivation. How does proper initiation and spreading of Xist along the X chromosome occur? The identification of regulatory factors uniquely interacting with the silent X chromosome at various steps may also provide much needed insight into the XCI mechanism.
While the mechanism of XCI is not completely defined, Xist is one of the very well-characterized regulatory factors central to XCI. Xist is a 17kb long non-coding RNA (lncRNA) whose expression is very tightly regulated in a cell-stage specific manner. The stable monoallelic Xist expression marks the initiation of the silencing of Xi and coats the chromosome in cis[5]. Mouse embryonic stem cells (ESCs) have served as a model system to dissect the early steps of XCI, because mouse ESCs carry two active X chromosomes (Xa)[6,7]. But at the onset of differentiation, the Xist transcription is upregulated on Xi along with several other stochastic fluctuations in regulatory factors, such as pluripotency factors and epigenetic regulators[8]. While Xist is essential for the initiation of XCI, an antisense non-coding RNA, Tsix plays a crucial role in preventing Xist expression from the Xa[9]. Although the mechanistic details of Tsix-mediated Xist down-regulation are not completely understood, a recent study showed that the Hedgehog paracrine system induces Tsix expression in pluripotent stem cells[10]. Another study suggested that Xist down-regulation could result from either binding of transcriptional factors within Tsix, or from counter-current movement of a Polymerase complex that would inhibit production of a sense Xist transcript[9,11,12]. Alternatively, Tsix itself could be a functional repressor by sequestering Xist and targeting Xist for degradation by RNA interference (RNAi) dependent mechanisms[12]. Once XCI is established, Tsix is only transcribed at low level from the Xa. Additional repressors of Xist expression include the pluripotency factors, such as SOX2, OCT4, NANOG, and PRDM14, which prevent XCI in undifferentiated ESCs[13]. Down-regulation of the pluripotent factors precedes the initiation of XCI. Several positive X-encoded activators of Xist expression have also been identified and includes, protein coding genes (Rnf12)[14], and ncRNAs (Ftx and Jpx)[15,16]. The expression of several of these factors is upregulated in the early stages of differentiation, explaining, at least in part, their involvement in XCI.
The next wave of XCI is the establishment of silencing on Xi, which entails multiple steps, ensuing with the spreading and coating of the Xist on Xi. While the active recruitment of silencing machinery to Xi is a key component of establishing XCI, well-defined structural domains of Xist are also crucial for its function. The structural domains of Xist could instruct the Xist:chromatin interactions and direct the subsequent assembly of the epigenetic silencing machinery on Xi. One of the crucial regulatory region is the X inactivation center (Xic), which is enriched in protein-coding genes, non-coding genes, and regulatory elements important for XCI[17]. It is likely that the order of individual gene silencing on Xi is dictated by physical gene location relative to the Xic. Additionally, there are several tandem repeats present in the Xist RNA that are described as repeats A-F[18]. Repeat A region of Xist, located in a small internal transcript (RepA), has emerged as a key player in initiating the silencing events on Xi[19]. Additionally, the repeat C region of Xist interacts with transcription factor, YY1 and this interaction is essential to tether Xist to the nucleation center on Xi[20]. Another important step in the stabilization of Xi is the Xist-mediated recruitment of chromatin modifiers, including Polycomb repressive complexes 1 and 2 (PRC1 and PRC2) and macroH2A[19,21,22]. These proteins alter the epigenetic landscape of Xi by introducing repressive marks such as H3K27me3, H3K9me2, H4K20me1, and H2Aub1, which are essential for stabilizing the silencing of Xi[23-26][Figure 1]. Although several studies provide substantive evidence to support the role of PRC in XCI, but the mechanism of PRC-Xist interaction has been a topic of debate. Early experiments demonstrated a PRC2 interaction with RepA region of Xist, which was then followed by PRC1 recruitment[27]. However, more recently, several studies pointed to more direct role of PRC1 in XCI, thereby pointing to a non-canonical recruitment of PRC to Xi[28]. Some of these differences can be attributed to cell lineages used in the different studies or the technical approaches; however more in depth investigation is needed to better understand the PRC-mediated silencing of Xi. The discrepancies in the conclusion of various studies regarding the role of PRC in XCI have been discussed in details elsewhere[29] and are not discussed further in this review. DNA methylation has also been found to play an important role in the stabilization of the Xi state[30]. In summary, the plethora of epigenetic modifications accumulating on Xi define its higher-order organization, which is essential for stabilizing the three dimensional structure of Xi[31].
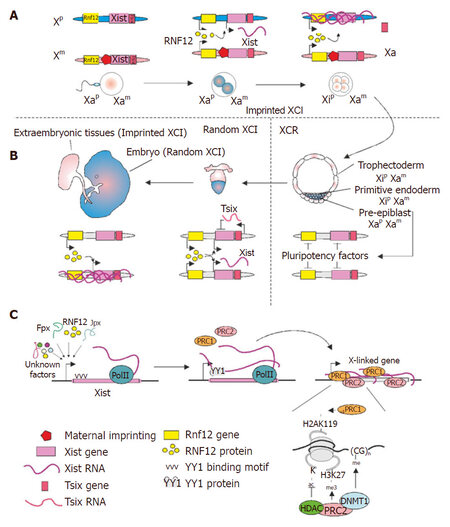
Figure 1. Model of X chromosome inactivation (XCI) in female embryos. (A, B) Imprinted and random XCI during development. XCI occurs in two distinct Xist-dependent waves. (A) The first phase is the imprinted XCI that commences at the two-to-four cell stages. Cells in the female embryo initiate the imprinted XCI that selectively inactivates the Xist coated paternal X chromosome (Xp). Xist expression from the maternal X chromosome (Xm) is inhibited due to the imprint signatures. RNF12 is a trans-activator that functions in the imprinted XCI in a dose-dependent manner. (B) As the embryo further develops into a blastocyst, Xp is reactivated in the pre-epiblast cells of inner cell mass (ICM). Subsequent initiation of random XCI (rXCI) occurs in ICM right after implantation and randomly affects either the Xp or Xm. Inactive state is stably maintained through further mitosis in the soma cells. Two copies of Rnf12 were also suggested to be necessary to activate Xist during random XCI. (C) Model of the sequence of events leading to silencing of X chromosome. Random XCI has three well-demarcated stages: initiation, establishment, and maintenance. Upon initiation Xist is regulated by several cis and trans-regulatory factors. Xist up-regulation in cis is followed by its tethering to the nucleation center by YY1, silencing then further spreads on the entire X chromosome. Xist recruits Polycomb repressive complexes (PRC 1 and 2), histone deacetylase 3 (HDAC3), and DNA (cytosine-5)-methyltransferase 1 (DNMT1), which mediate methylation of H3K27, histone deacethylation and CpG islands methylation respectively
Previously, XCI has been considered unidirectional during later stages of development, but more recently, the idea of reactivating Xi has emerged. Evidence suggests that the epigenetic state of Xi can be reversed by genetic or pharmacological manipulations[32-34]. Although these results were paradigm shifting, they were not unexpected due to the inherent ability of Xi to shuttle between the active and inactive states during embryogenesis. During embryo formation, XCI occurs in two distinct stages: first, imprinted inactivation of paternal X (Xp), and second, random inactivation of either paternal (Xp) or maternal X (Xm) upon differentiation[35,36]. Xp is inactivated shortly after fertilization due to maternal imprint, but it is subsequently reactivated at the blastocyst stage of embryo development. This process is followed by random XCI in the cells of the inner cell mass (ICM) of the embryo, and maintained throughout mitosis[37]. Extensive analysis using in vitro differentiation of cells isolated from ICM has enabled a comprehensive understanding of the early events in the random XCI [Figure 1]. Further, the analysis of XCI status in epigenetically stable basal breast cancer and ovarian cancer cell lines have revealed defects in Xi, evident by cells carrying two Xa[38]. Although a preferential loss of Xi and gain of an additional copy of Xa could explain the Xa polyploidy, the reactivation of Xi could not be ruled out. In fact, by identifying single nucleotide polymorphism (SNP) differences in the X-linked genes, it was demonstrated that Xi was indeed reactivated in a subset of breast cancer cells[39]. Together, these findings point to the tantalizing possibility of synthetically manipulating the state of Xi, which could have far reaching therapeutic implications for monogenic X-linked diseases, such as Rett syndrome, CDKL5 syndrome, and Fragile X syndrome. The reversal of XCI could compensate for the gene deficiencies by expressing the endogenous wild-type copy from Xi. Several studies have demonstrated that upon the interference with the function of regulatory factors of XCI, the Xi can be reactivated in mouse embryonic fibroblasts, fibroblast cell lines and cortical neurons[32-34]. However, before embarking on a translational approach based on the idea of X chromosome reactivation (XCR), a detailed understanding of the XCR mechanism and regulatory factors is warranted.
Here, we review recent advancements in the XCI field that has provided crucial insights into the mechanism of XCI and the regulators of the epigenetically inert landscape of Xi. We also discuss different methodologies that combine biochemical, genomics, genetic and proteomics strategies to investigate the mechanism of XCI. Using these new multidisciplinary technological strategies, we are better equipped with tools that can guide the reversal of the established inactive state of the X chromosome. Identification of XCI factors (XCIFs) and their individual roles in the silencing process are essential for comprehension of the mechanism as a whole and could potentially aid development of novel approaches to treat X-linked diseases.
Recent technological advances providing new mechanistic insights into the XCI
A high level of epigenetic stringency of XCI accentuates the fascination with this paradigmatic phenomenon, but at the same time its complexity has contributed to the stalled progress. The silencing of mammalian X chromosome requires a well-regulated cascade of chromatin changes that are orchestrated by a battery of XCI regulators, which include both cis and trans-acting protein-coding as well as non-coding RNAs. Several of these crucial regulators remain unknown, mainly due to technical difficulties in discriminating molecular events unique to Xi; for example specific and non-specific binding interactions, gene expression, and epigenetic modifications from two identical X chromosomes and binding partners of the 17kb long Xist transcript. Even with the development of more unbiased and sensitive methods, the variations between applied protocols and chosen biological systems have resulted in non-overlapping and often inconclusive results. Here, we discuss recent technological developments that have expedited the identification of XCI regulators and have provided intricate molecular details of initiation, maintenance, and establishment of XCI.
Loss-of-function genetic screens
A candidate-based approach is not the most feasible and efficient method of defining the mechanism of XCI. But lack of tools to interrogate this question on the genome-wide scale has been a limiting factor for a very long time. In last few decades, the genetic screens combined with high-throughput technologies have revolutionized the field by the identification of regulatory factors functioning in the epigenetic silencing mechanism and ordering them into a pathway. The two widely famous genetic screening strategies include gain-of-function screens that involve ectopic expression of genes using complementary DNA (cDNAs) and loss-of-function screens that target the transcriptome using RNAi technology. Although no gain-of-function screen has been done to investigate XCI, recently several loss-of-function screens have dissected the mechanism of X chromosome silencing [Figure 2]. Many of these large-scale screens used a mouse fibroblast cell lines carrying transgenic reporter on Xi (GFP[32,33,40] and luciferase[34]), to determine the efficiency of Xi reactivation. Quite a few screens identified XCIFs that overlap between these screens, thereby validating the findings and increasing confidence in the RNAi-based screening tools[32-34]. The success of these initial screens also encouraged the use of more focused libraries, such as the chromatin modifiers[41] and RNA-binding proteins[42]. While shRNA-based libraries have immensely accelerated the field, use of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-based libraries is expected to fast-track our understanding of the mechanisms of XCI. This is mainly because CRISPR technology has a capacity to cause complete deletions of genes that will be more powerful in identifying XCI-specific genes and in ruling out genes that are essential for survival.
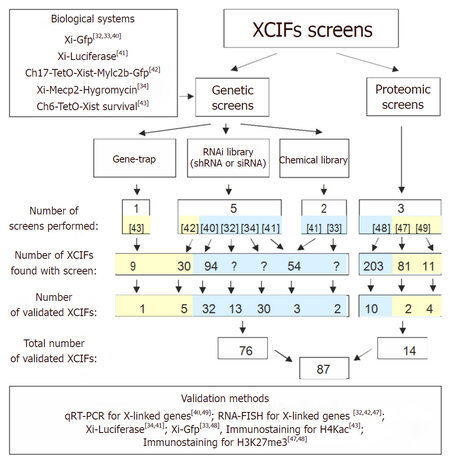
Figure 2. Strategies used to search for new X chromosome inactivation (XCI) factors (XCIFs) in all 10 screens performed. Seven genetic screens[32-34,40-43] and three proteomics screens[47-49] identified more than 500 XCIFs. So far, 87 of them have been validated using candidate approaches. Screens highlight in yellow were performed in embryonic cells and focus on establishment of XCI, screens highlight in blue were performed in differentiating cells and focus on maintenance of XCI and possibility of X chromosome reactivation (XCR)
An important criterion of successful screening strategy format is a biological phenotype that provides a powerful readout for evaluating the reactivation of Xi. In this recombination era, there has been a surge in the development of very useful reporter in vitro tools to study XCI, including immortalized cell lines with Xi-linked gene tagged to reporters[34,41] and mouse embryonic stem cells[43]. With several mouse models available, an in vivo screen may provide more therapeutically relevant gene targets. The availability of these tools could also facilitate phenotypic screens, which would identify factors that are selective to XCI initiation, maintenance, or establishment.
Chemical based genetics screens
Another unbiased approach to identify X chromosome regulatory factors relies on small molecule perturbations. While chemical screens could reveal tractable molecular targets functioning in XCI, they could also provide an efficient tool to probe the targets’ functions and outline their mechanism-of-action. As depicted in Figure 2, the workflow of a chemical screen resembles that of a genetic screen and relies on a phenotypic screening to identify the small molecule(s) that gives a desired phenotype. In the case of XCI, this phenotype is measured by the activation of a reporter gene. Recently a compound screen using ~400,000 molecules identified synergism between DNA methylation and Aurora kinase pathways for XCR[33]. Another screen with a smaller library of ~4300 chemical inhibitors identified RRM2 inhibitors that enhance DNA demethylation by altering nucleotide metabolism causing XCR[41]. With an increasing repertoire of available small molecule libraries, chemical screening holds a great promise to complement classical genetics in discovering the regulatory factors involved in XCI.
RNA:protein interaction coupled to proteomics screens
A mammalian genome encodes several lncRNAs that associate with proteins to execute complex structural and regulatory functions. Xist is one of the paradigmatic lncRNAs, and deciphering its interacting regulatory functions is central to understanding the Xist-mediated epigenetic silencing of the X chromosome. Several tools are available to interrogate the lncRNA:protein interaction from a protein-centric view, but fewer methods have been available with lncRNA in focus. However, evolution in the field of immobilized oligonucleotide probes, affinity aptamers, and in vitro transcribed RNAs has allowed the capture of Xist:protein interactions in vivo[44,45]. One such assay is an extension of comprehensive identification of RNA-binding proteins by high-throughput sequencing (CHIRP-seq[46]) that uses a proteomics approach to identify the Xist-bound proteome, called CHIRP-MS[47]. CHIRP-seq is a an affinity-based method that generates high resolution map of lncRNA binding sites by capturing lncRNA:chromatin complexes using tiling antisense oligos.
Another method is identification of direct RNA interacting proteins (iDRiP) that captures Xist-interacting proteins using cDNA probes for Xist[48]. However, to minimize the background and increase the specificity, iDRiP method requires a cross-linking step and an additional DNAse treatment. Similarly, an RNA antisense purification-mass spectrometry (RAP-MS) method modified previous assays and captured Xist:protein complexes by using long antisense biotinylated DNA probes[49].
Additionally, new methods have been developed to interrogate the spatial organization of Xi in the nuclear 3D space and are discussed in details elsewhere[50]. An imortant example is capture hybridization analysis of RNA targets combined with high-throughput sequencing (CHART-seq). This approach revealed high-resolution maps of Xist interaction with chromatin across a developmental time course and pointed to the preferential Xist-binding to gene-rich islands before spreading to gene-poor domains[51].
Novel XCI players identified through unbiased genetic and proteomic screens
Since the discovery of Xist in the early 1990s, Xist has garnered great interest, but molecular details about its function in XCI have been limited. Coating by Xist precedes the silencing of Xi and initiates a cascade of events that are necessary for the maintenance and establishment of the silencing. Several of these functions are attributed to the characteristic molecular domains of Xist that include an abundance of repetitive sequences. It has also been speculated that Xist interacts with different regulators, a few of which have been identified by targeted RNA immunoprecipitation (RIP) assay. These regulators include ATRX, YY1, hnRNPU/SAF-A, and PRC2. YY1 and hnRNPU facilitate the recruitment of Xist to Xi during the initiation and maintenance phase of XCI respectively. YY1 is a transcription factor that possesses bivalent capacity to bind both DNA and RNA, thereby functioning to dock Xist to the chromatin[20]. The YY1 binding domain in Xist has been mapped to the Repeat C region using targeted RIP and electrophoretic mobility shift assays (EMSA)[20]. Another Xist-interacting protein crucial for its localization and identified through multiple screens is a matrix protein called hnRNPU[42,47,49]. Investigation of the hnRNPU by mutational analysis revealed arginine-glycine-glycine (RGG) motifs essential for its interaction with Xist[52]. ATRX, a high-affinity RNA-binding protein (RBP) directly interacts with RepA/Xist RNA and promotes loading of PRC2[27]. Without ATRX, PRC2 cannot interact with Xist or spread in cis along the X chromosome[53]. There is no denying the fact that use of targeted approaches have identified several key players in XCI, as outlined above; however, this progress has been slow. Recent technological advancements have revolutionized the discovery of new regulatory factors and provided detailed information to order them in a sequential pathway. Here, we have summarized different XCIFs discovered in recent years, based on their function in the silencing of the mammalian X chromosome.
Xist interacting proteins
Recently, three different groups used distinct methodologies to capture proteins that interact with Xist. These studies used RNA-centric approaches, including ChIRP-MS[47], iDRiP[48], and RAP-MS[49] to identify Xist interactome. All together, ~300 proteins have been identified to interact with Xist[Figure 2]. The screens identified several previously characterized Xist-interacting proteins, including hnRNPU, hnRNPK, AURKB, RAD21, CTCF, SPEN, and HDAC3, but the other known XCIF, like EZH2 and ATRX were not identified. Interestingly, the three screens lacked significant overlap between the screens that could be attributed to technical differences; for example, the efficiency of crosslinking or to the cell type used in these studies. A more directed functional validation across different cellular systems could further address these concerns. In the next section, we discuss some of the protein partners for Xist that have emerged from these screens and have been functionally validated [Table 1].
List of XCIFs identified to-date using genetic, biochemical and proteomics screens[32-34,40-43,47-49]
| Function | XCIF | Pharmalogical inhibitorstested (names)/availability (+/−) | Surface protein | Study | Chromosome mouse (human) |
|---|---|---|---|---|---|
| Cell cycle regulation | 53BP1 | − | − | [40] | 2 (15) |
| ANAPC5 | − | − | [40] | 5 (12) | |
| AURKA | VX680, MLN8237/+ | − | [32-34] | 2 (20) | |
| AURKB | VX680, MLN8237/+ | − | [33,49] | 11 (17) | |
| BRCA2 | − | − | [34] | 5 (13) | |
| CDC25a | − | − | [40] | 9 (3) | |
| CDK4 | + | − | [40] | 10 (12) | |
| CLASPIN | − | − | [34] | 4 (1) | |
| GINS3 | − | − | [34] | 8 (16) | |
| LRWD1 | − | − | [40] | 5 (7) | |
| MCM8 | − | − | [34] | 2 (20) | |
| NF1 | − | − | [32] | 11 (17) | |
| ORC2 | − | − | [40] | 1 (2) | |
| PARP6 | + | − | [34] | 9 (15) | |
| PLK2 | + | − | [34] | 13 (5) | |
| RAD21 | + | − | [34,49] | 15 (8) | |
| SKP2 | + | − | [40] | 15 (5) | |
| SYCP1 | − | − | [40] | 3 (1) | |
| ZWINT | − | − | [40] | 10 (10) | |
| RNA binding and procesing | BCAS2 | − | − | [40] | 3 (1) |
| LBR | − | − | [49] | 1 (1) | |
| METTL3 | + | − | [40] | 14 (14) | |
| PTBP1 | − | − | [40] | 10 (19) | |
| RBM15 | − | − | [42] | 3 (1) | |
| RRM2 | HU, Resveratrol/+ | − | [41] | 12 (2) | |
| SSB | − | − | [40] | 2 (2) | |
| WTAP | − | − | [42] | 17 (6) | |
| HNRNPK | − | − | [42,47] | 13 (9) | |
| HNRNPU | − | − | [42,47,49] | 17 (1) | |
| Signaling kinases | CCND2 | + | − | [40] | 6 (12) |
| ERBB4 | + | − | [34] | 1 (2) | |
| PDPK1 | OSU-04402, BX912/+ | − | [32,34] | 17 (16) | |
| PIK3CB | LY294002; GNE-407 | − | [32,34] | 9 (3) | |
| PRKD3 | + | − | [34] | 17 (2) | |
| PRPF4B | + | − | [40] | 13 (6) | |
| SGK2 | + | − | [34] | 2 (20) | |
| SRPK2 | + | − | [40] | 5 (7) | |
| TGF-B/BMP signaling | ACVR1 | + | + | [32,34] | 2 (2) |
| ACVR1b | + | + | [34] | 15 (12) | |
| Bmpr2 | + | + | [34] | 1 (2) | |
| PRRX1 | − | − | [40] | 1 (1) | |
| RNF165 | − | − | [32] | 18 (18) | |
| SMAD2 | − | − | [34] | 18 (18) | |
| SNIP1 | − | − | [34] | 4 (1) | |
| ZFYVE9 | − | − | [34] | 4 (1) | |
| Transcriptional regulation | ATF7IP | − | [41] | 6 (12) | |
| ARID4B | − | − | [34] | 13 (1) | |
| CTCF | − | − | [49] | 8 (16) | |
| DNMT1 | 5-Azacytidine/+ | − | Multiple studies | 9 (19) | |
| DTX3L | − | − | [34] | 16 (3) | |
| H2AFX | − | − | [40] | 9 (11) | |
| HMGN3 | − | − | [40] | 9 (6) | |
| ID3 | − | − | [40] | 4 (1) | |
| KLF17 | − | − | [40] | 4 (1) | |
| MBD1 | − | − | [40] | 18 (18) | |
| MSL2L1 | − | − | [34] | 9 (3) | |
| NIPBL | − | − | [34] | 15 (5) | |
| PBRM1 | + | − | [40] | 14 (3) | |
| PHC1 | − | − | [40] | 6 (12) | |
| SOX5 | − | − | [32] | 6 (12) | |
| SPEN | − | − | [42,43,47,49] | 4 (1) | |
| TITIN (TTN) | − | + | [34] | 2 (2) | |
| TOP1 | + | − | [49] | 2 (20) | |
| TOP2A | + | − | [49] | 11 (17) | |
| YAF2 | − | − | [40] | 15 (12) | |
| YY1 | − | − | [12] | 12 (14) | |
| ZBP1 | − | − | [40] | 2 (20) | |
| ZNF496 | − | − | [32] | 9 (19) | |
| WNT signaling | DVL2 | + | − | [40] | 11 (17) |
| HDAC3 | TSA/+ | − | [34,40,49] | 18 (5) | |
| MYC | + | − | [40] | 15 (8) | |
| PYGO1 | − | − | [32] | 9 (15) | |
| SMARCA4 | − | − | [49] | 9 (19) | |
| SMARCD2 | − | − | [34] | 11 (17) | |
| Other | ACACA | + | − | [34] | 11 (17) |
| ACP1 | − | − | [40] | 12 (2) | |
| ACSS1 | − | − | [34] | 2 (20) | |
| BCAS1 | − | − | [40] | 2 (20) | |
| C17ORF98 | − | − | [32] | 11 (17) | |
| CML3 | − | − | [40] | 6 (4) | |
| FBXO8 | − | − | [32] | 8 (4) | |
| LAYN | − | − | [32] | 9 (11) | |
| PSMC4 | − | − | [40] | 7 (19) | |
| SMAC1A | − | − | [49] | X (X) | |
| SMC3 | − | − | [49] | 19 (10) | |
| STC1 | − | − | [32] | 14 (8) | |
| SUN2 | − | − | [49] | 15 (22) |
SPEN (also known as SHARP or Mint) was identified through both proteomics and genetic screens and belongs to the Split End RNA binding protein (SPEN) family of transcriptional repressors. SPEN proteins execute transcriptional regulatory functions via three copies of RNA recognition motifs (RRMs) and ortholog C-terminal (SPOC) domains[54]. SPEN is a part of multi-subunit complex that consist of NCoR/SMRT co-repressor complex required for its interaction with HDAC3. Additionally, SPEN interacts with a regulatory ncRNA, SRA, and this interaction modulates nuclear hormone receptor signaling[55]. McHugh et al.[49] found that knockdown of NCoR/SMRT or HDAC3 abrogates Xist-mediated silencing. Further, transcriptome analysis of X-linked genes in ESCs expressing functionally defective SPEN abrogated Xist ability to initiate gene repression[43]. Identical results were obtained in multiple reporter systems, and showed an increase in the expression of X-linked genes, upon RNAi-based Spen depletion[42,47,49]. Although the X-linked gene expression was affected by the inhibition of SPEN function, surprisingly Xist coating of Xi was not affected[43]. This suggested that SPEN is not crucial for Xist localization, accumulation, or spreading but essential for Xi-linked gene silencing. Subsequent studies by Chu et al.[47] and Monfort et al.[43], mapped SPEN binding to the A-repeat of Xist. It has been suggested that SPEN could create the initial silenced compartment on the X chromosome by excluding Pol II, providing an Xi silencing permissive environment. Evidently, Pol II was found to be localized in the Xist clouds of SPEN depleted ESCs, and loss of SPEN altered the recruitment of chromatin modifiers, like PRC2 complex and Ring1b[49]. Interestingly, similar analysis by Monfort et al.[43] failed to reproduce the complete loss of Polycomb complexes (EZH2 and RING1B proteins), but the reported partial loss was sufficient to de-repress X-linked genes in the SPEN depleted ESCs. These contradictory results remain to be addressed, and the exact Xist-independent mechanism by which SPEN regulates XCI is unclear.
RBM15 and WTAP (Wilms tumor-assosiated protein) are two proteins recently linked to the N6-adenosine (m6A) RNA methyltransferase complex. RBM15 is an RNA binding protein (RBP) and a SPEN family member, containing a SPOC domain; however, it does not have redundant functions with SPEN. Although several region of Xist are m6A modified but the functional significance of this post-transcriptional modification for XCI remains unclear. It is suspected to mediate RNA splicing or RBP binding. RBP15 along with a very similar protein RBM15B and methyltransferase-like 3 (METTL3) facilitates the recruitment of m6A machinery to Xist in a WTAP-dependent manner[56]. RMB15 was identified as a Xist-interacting protein through iDRiP[42], and a proteomic study confirmed its interaction with WTAP[57]. Xist interaction with RMB15-WTAP complex was mapped to A-repeat and implicated in transcriptional silencing of the X chromosome. Further, a study by Moindrot et al.[42] showed that knockdown of RBM15 and WTAP increased the number of cells with biallelic signals for X-linked genes, Pgk1 and Rnf12, demonstrating transcriptional reactivation of Xi-linked genes. Interestingly, like SPEN, the loss of WTAP and RBM15 did not interfere with Xist recruitment on Xi[42].
hnRNPK is an RBP and a member of the hnRNP protein family. Another member of this family is hnRNPU, is a Xist-interacting protein, and loss of hnRNPU reactivated several Xi-linked genes[58]. Along with hnRNPU, hnRNPK was one of the most abundant proteins identified in a ChIRP-MS screen carried out using cells with doxycycline-inducible Xist on chromosome 11[47]. This protein was identified through multiple screens using different cell lineages; epiblast stem cells (EpiSCs) that had just undergone random XCI and trophoblast stem cells where Xp is always silenced[47]. The reactivation of Xi-linked Usp9x in hnRNPK-depleted EpiSCs, confirmed that hnRNPK regulates XCI[47]. Surprisingly, northern blot analysis showed that the depletion of hnRNPK does not impact Xist abundance or splicing[47]. As with SPEN, hnRNPK depletion did not alter Xist localization on Xi, however PRC2 recruitment was significantly decreased. Chu et al.[47] also showed that hnRNPK directly binds Xist, with its strongest interaction located downstream of Repeat F.
Chromatin modifiers identified through the screens
The global state of chromatin undergoes extensive remodeling, causing transition from a relatively open configuration to a more compact state. SWI/SNF is a part of the chromatin-remodeling complex found in all living organisms. In eukaryotes, this complex is capable of altering the position of nucleosomes along DNA, which considerably impacts transcription[59]. SWI/SNF is also a platform that interacts with many transcription factors and chromatin remodeling proteins. A few components of this complex were found to be important for XCI, including SMARCA4[48] and SMARCD2[34]. It is suggested that SWI/SNF interaction with Xist is required for proper maintenance of PRC2 function on the Xi[48].
Transcriptional regulators of XCI
There are number of cis-acting regulatory elements that are located in the Xic. Several studies have used genetic approaches to delineate their roles. Identified factors include non-coding RNA genes Jpx[16], Linx[60], Tsx[61], Xite[62], and Ftx[15] as well as protein-coding genes Rnf12[14], Chic1[17], Ppnx[63], Xpr[64], and Nap1L2[63]. With few exceptions, the exact mechanism by which these factors modulate Xist or Tsix function remains to be determined. While interrogation of cis-regulation of XCI continues, the trans-regulation of this process has also garnered immense interest. Trans-regulation of XCI refers to the regulation of X-linked gene expression by diffusible factors that can either activate or inhibit the silencing of Xi. As discussed earlier, pluripotency factors are one of the trans-acting negative regulators of XCI[65].
Recently, Bhatnagar et al.[32] and Sripathy et al.[34] have carried out unbiased large-scale genome-wide screens to identify cis and trans-acting regulators of XCI. Both the screens used fundamentally different reporters to identify Xi reactivators but identified several XCIFs in common [Table 1 and Figure 3]. Among identified XCIFs there is a large representation of BMP/TGF-β signaling pathway components found in three different studies[32,34,40] [Table 1 and Figure 3]. Inhibition of this pathway was shown to decrease Xist expression. Interestingly, Xist promoter contains several SMAD-binding motifs in close proximity to the YY1-binding site crucial for anchoring Xist to Xi. This suggests that several regulatory pathways may converge to stringently regulate Xist expression. Additionally, RNF12 also controls SMAD transcription factor levels, a process that may function as a potential feedback loop for regulating Xist levels[66].
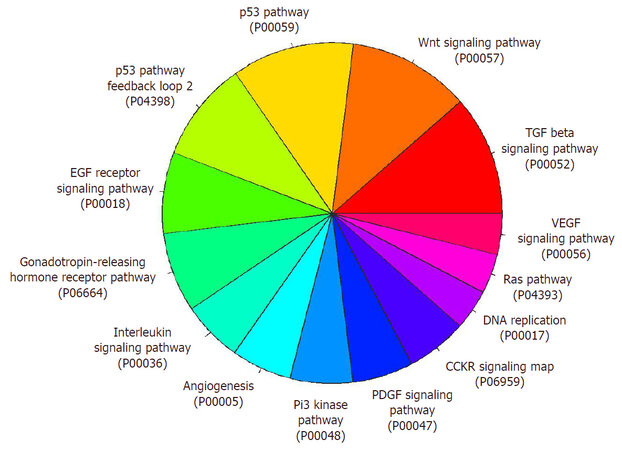
Figure 3. Functional annotation clustering of X chromosome inactivation (XCI) factors (XCIFs). Pathway based evaluation for 87 XCIFs identified through multiple genetic and proteomics screens were categorized according to biological process and molecular function using PANTHER protein class ontology
Additionally, the identification of XCIFs raises the prospects of Xi reactivation as a therapeutic strategy due to several reasons. First, several of the XCIFs identified through the screens possess enzymatic activity that can be targeted by pharmacological agents, for instance DNMT1 and RRM2. Secondly, another major category of XCIFs identified includes serine/threonine kinases, such as for example, PI3kinase signaling network (PDPK1, SGK2 and PRKD3), ACVR1 receptor, DNMT1 and Aurora kinase A, which can be targeted by several small molecule inhibitors.
The 3D conformation of the Xi
The X chromosome was originally identified as a Barr body due to its peculiar heterochromatin structure in the nucleus. Classical cytological and electron microscopy studies have revealed a unique ultrastructure as well as a distinctive localization of Xi. The advent of 3C technologies and the use of DNA FISH combined with super-resolution microscopy have shed light on the organization of the X chromosome. Chromosome Conformation Capture combined with High-throughput sequencing (Hi-C) showed that Xi has unique structural features that dictate its conformation and localization. Xi is condensed and partitioned into extremely large loops, up to 77 megabases long, called "superloops” that are organized into two macrostructures, called "superdomains”. One of the key regulators of Xi organization is the macrosatellite repeat locus Dxz4, which encodes two lncRNAs[67] and separates the “superdomains”. Although the mechanism by which Dxz4 regulates the organization and 3D structure of Xi warrants more investigation, several new functions for Dxz4 have emerged from recent studies. First, Dxz4 could sequester the nucleophism, a component of the nucleolus essential for the formation of nucleolus-associated domains (NADs)[68]. These NADs could influence positioning of Xi in a nucleus. Secondly, Dxz4 harbors lncRNA that includes CTCF binding, which is essential for XCI initiation[67]. Thirdly, Dxz4 plays a role in mediating long-range intrachromosomal interactions on the Xi[69]. Importantly, the deletion of Dxz4 from Xi leads to the disappearance of superdomains and superloops[70], but does not interfere with XCI initiation or with the enrichment of epigenetic marks[70].
Recently, a 3D map of Xi revealed that the formation and progression of the loops brings the distant regions of Xi together, affecting their transcriptional status[71]. One such example is the “escape genes” that are in close vicinity of one another, even if they are physically separated by megabase pair distance on Xi[70,71]. Existence of mechanisms regulating escape from XCI suggests that this process is not only an imperfection of the natural system, but that it could be very well-orchestrated with important biological implications.
Final remarks
XCI is a complex mechanism that requires both cis and trans-acting regulatory factors for the immaculate execution of the transcriptional silencing of Xi. Despite the discovery of XCI over 50 years ago, several open questions remain. How many XCI activators and repressors exist? How do these regulators function in different stages of XCI, and are they cell-type or cell-stage specific? These and many other questions remain to be addressed. But as futurists predicted, we are in a state of active technological revolution; this advancement has provided us with powerful tools to interrogate the mechanism of XCI much more intricately than what was possible only a few years ago. Recent studies have identified several regulatory elements that are required for the initiation, establishment, and maintenance of XCI. Such studies have also revealed the plasticity of the inactive state of the X chromosome, which was previously limited to early stages of embryogenesis or induced pluripotency.
As discussed earlier, new tools have been developed recently that have aided our understanding of XCI, especially by identifying proteins that either interact with Xist or transcriptionally regulate its expression. Using elegant tools, the structural and organizational maps of Xi in the nucleus have also been generated. In light of recent findings, we now realize that XCI is more variable than previously thought, and that its outcome is dependent on the crosstalk of different regulatory elements at multiple levels. Diverse molecular, biochemical, and genetic approaches have identified more than 500 proteins that are involved in XCI [Figure 2], with 87 of them having been validated using a single-gene approach [Table 1]. How these different proteins regulate the silencing of XCI, or which steps of XCI are regulated by these factors, remains to be addressed. However, the emerging fact from these studies does point to multiple gene-silencing mechanisms. Pathway analysis[72,73] revealed that newly discovered XCIFs can be categorized into 14 different pathways, that include: Wnt signaling pathway, p53 pathway, TGF-β signaling pathway, p53 pathway feedback loops 2, and EGF receptor signaling pathway [Figure 3].
Finally, an important therapeutically relevant question is, can Xi be reactivated to compensate for gene deficiencies in X-linked diseases? One such example is Rett syndrome, which is a rare neurodevelopmental disease caused by loss-of-function mutations in the X-linked gene MECP2 encoding Methyl CpG binding protein. As discussed above, reactivating Xi-linked MECP2 in Rett syndrome patients can restore symptoms associated with the disease and possibly reverse the disease[74-76]. But the lack of understanding of XCI has limited its applicability as a target for treatment. The identification of molecular players in XCI could be beneficial in expediting efforts to develop XCR-based therapeutic approaches. In fact, many of the XCIFs identified are druggable and efforts are underway to target them for pharmacological interventions.
Declarations
Authors' contributionsAll authors contributed to the paper writing .
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
2. Ercan S, Lieb JD. C. elegans dosage compensation: a window into mechanisms of domain-scale gene regulation. Chromosom Res 2009;17:215-27.
3. Hallacli E, Akhtar A. X chromosomal regulation in flies: when less is more. Chromosom Res 2009;17:603-19.
4. Maduro C, de Hoon B, Gribnau J. Fitting the puzzle pieces: the bigger picture of XCI. Trends Biochem Sci 2016;41:138-47.
5. Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature 1996;379:131-7.
6. Pintacuda G, Cerase A. X inactivation lessons from differentiating mouse embryonic stem cells. Stem Cell Rev Reports 2015;11:699-705.
7. Shen Y, Matsuno Y, Fouse SD, Rao N, Root S, Xu R, Pellegrini M, Riggs AD, Fan G. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc Natl Acad Sci U S A 2008;105:4709-14.
8. Chaumeil J, Okamoto I, Guggiari M, Heard E. Integrated kinetics of X chromosome inactivation in differentiating embryonic stem cells. Cytogenet Genome Res 2002;99:75-84.
9. Stavropoulos N, Lu N, Lee JT. A functional role for Tsix transcription in blocking Xist RNA accumulation but not in X-chromosome choice. Proc Natl Acad Sci U S A 2001;98:10232-7.
10. Del Rosario BC, Del Rosario AM, Anselmo A, Wang PI, Sadreyev RI, Lee JT. Genetic intersection of Tsix and Hedgehog signaling during the initiation of X-chromosome inactivation. Dev Cell 2017;43:359-71.e6.
11. Lee JT, Davidow LS, Warshawsky D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 1999;21:400-4.
12. Shibata S, Lee JT. Characterization and quantitation of differential Tsix transcripts: implications for Tsix function. Hum Mol Genet 2003;12:125-36.
13. Payer B, Lee JT. Coupling of X-chromosome reactivation with the pluripotent stem cell state. RNA Biol 2014;11:798-807.
14. Barakat TS, Gunhanlar N, Pardo CG, Achame EM, Ghazvini M, Boers R, Kenter A, Rentmeester E, Grootegoed JA, Gribnau J. RNF12 activates Xist and is essential for X chromosome inactivation. PLoS Genet 2011;7:e1002001.
15. Chureau C, Chantalat S, Romito A, Galvani A, Duret L, Avner P, Rougeulle C. Ftx is a non-coding RNA which affects Xist expression and chromatin structure within the X-inactivation center region. Hum Mol Genet 2011;20:705-18.
16. Tian D, Sun S, Lee JT. The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 2010;143:390-403.
17. Augui S, Nora EP, Heard E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat Rev Genet 2011;12:429-42.
18. Brown CJ, Hendrich BD, Rupert JL, Lafrenière RG, Xing Y, Lawrence J, Willard HF. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992;71:527-42.
19. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008;322:750-6.
21. Plath K, Talbot D, Hamer KM, Otte AP, Yang TP, Jaenisch R, Panning B. Developmentally regulated alterations in Polycomb repressive complex 1 proteins on the inactive X chromosome. J Cell Biol 2004;167:1025-35.
22. Schoeftner S, Sengupta AK, Kubicek S, Mechtler K, Spahn L, Koseki H, Jenuwein T, Wutz A. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J 2006;25:3110-22.
23. Costanzi C, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 1998;393:599-601.
24. Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL. Methylation of histone H3 at Lys-9 Is an early mark on the X chromosome during X inactivation. Cell 2001;107:727-38.
25. Rougeulle C, Chaumeil J, Sarma K, Allis CD, Reinberg D, Avner P, Heard E. Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X chromosome. Mol Cell Biol 2004;24:5475-84.
26. Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science 2003;300:131-5.
27. Sarma K, Cifuentes-Rojas C, Ergun A, Del Rosario A, Jeon Y, White F, Sadreyev R, Lee JT. ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell 2014;159:869-83.
28. Almeida M, Pintacuda G, Masui O, Koseki Y, Gdula M, Cerase A, Brown D, Mould A, Innocent C, Nakayama M, Schermelleh L, Nesterova TB, Koseki H, Brockdorff N. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 2017;356:1081-4.
29. Brockdorff N. Polycomb complexes in X chromosome inactivation. Philos Trans R Soc B Biol Sci 2017;372:20170021.
30. Sharp AJA, Stathaki E, Migliavacca E, Brahmachary M, Montgomery SB, Dupre Y, Antonarakis SE. DNA methylation profiles of human active and inactive X chromosomes. Genome Res 2011;21:1592-600.
31. Cotton AM, Price EM, Jones MJ, Balaton BP, Kobor MS, Brown CJ. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum Mol Genet 2015;24:1528-39.
32. Bhatnagar S, Zhu X, Ou J, Lin L, Chamberlain L, Zhu LJ, Wajapeyee N, Green MR. Genetic and pharmacological reactivation of the mammalian inactive X chromosome. Proc Natl Acad Sci U S A 2014;111:12591-8.
33. Lessing D, Dial TO, Wei C, Payer B, Carrette LL, Kesner B, Szanto A, Jadhav A, Maloney DJ, Simeonov A, Theriault J, Hasaka T, Bedalov A, Bartolomei MS, Lee JT. A high-throughput small molecule screen identifies synergism between DNA methylation and Aurora kinase pathways for X reactivation. Proc Natl Acad Sci U S A 2016;113:14366-71.
34. Sripathy S, Leko V, Adrianse RL, Loe T, Foss EJ, Dalrymple E, Lao U, Gatbonton-Schwager T, Carter KT, Payer B, Paddison PJ, Grady WM, Lee JT, Bartolomei MS, Bedalov A. Screen for reactivation of MeCP2 on the inactive X chromosome identifies the BMP/TGF-β superfamily as a regulator of XIST expression. Proc Natl Acad Sci U S A 2017;114:1619-24.
35. Huynh KO, Lee JT. Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature 2003;426:857-62.
36. Okamoto I. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 2004;303:644-9.
37. van den Berg IM, Galjaard RJ, Laven JS, van Doorninck JH. XCI in preimplantation mouse and human embryos: first there is remodelling. Hum Genet 2011;130:203-15.
38. Sirchia SM, Tabano S, Monti L, Recalcati MP, Gariboldi M, Grati FR, Porta G, Finelli P, Radice P, Miozzo M. Misbehaviour of XIST RNA in breast cancer cells. PLoS One 2009;4:e5559.
39. Chaligné R, Popova T, Mendoza-Parra MA, Saleem MA, Gentien D, Ban K, Piolot T, Leroy O, Mariani O, Gronemeyer H, Vincent-Salomon A, Stern MH, Heard E. The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res 2015;25:488-503.
40. Chan KM, Zhang H, Malureanu L, van Deursen J, Zhang Z. Diverse factors are involved in maintaining X chromosome inactivation. Proc Natl Acad Sci U S A 2011;108:16699-704.
41. Minkovsky A, Sahakyan A, Bonora G, Damoiseaux R, Dimitrova E, Rubbi L, Pellegrini M, Radu CG, Plath K. A high-throughput screen of inactive X chromosome reactivation identifies the enhancement of DNA demethylation by 5-aza-2′-dC upon inhibition of ribonucleotide reductase. Epigenetics Chromatin 2015;8:42.
42. Moindrot B, Cerase A, Coker H, Masui O, Grijzenhout A, Pintacuda G, Schermelleh L, Nesterova TB, Brockdorff N. A pooled shRNA screen identifies Rbm15, Spen, and Wtap as factors required for Xist RNA-mediated silencing. Cell Rep 2015;12:562-72.
43. Monfort A, Di Minin G, Postlmayr A, Freimann R, Arieti F, Thore S, Wutz A. Identification of Spen as a crucial factor for Xist function through forward genetic screening in haploid embryonic stem cells. Cell Rep 2015;12:554-61.
44. Ferrè F, Colantoni A, Helmer-Citterich M. Revealing protein-lncRNA interaction. Brief Bioinform 2016;17:106-16.
45. Barra J, Leucci E. Probing long non-coding RNA-protein interactions. Front Mol Biosci 2017;4:45.
46. Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 2011;44:667-78.
47. Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY. Systematic discovery of Xist RNA binding proteins. Cell 2015;161:404-16.
48. Minajigi A, Froberg JE, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, Lee JT. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015;349:aab2276.
49. McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, Sweredoski MJ, Shishkin AA, Su J, Lander ES, Hess S, Plath K, Guttman M. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015;521:232-6.
50. Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol 2016;17:756-70.
51. Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg-Schoenberg M, Bowman SK, Kesner BA, Maier VK, Kingston RE, Lee JT. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 2013;504:465-9.
52. Kiledjian M, Dreyfuss G. Primary structure and binding activity of the hnRNP U protein: binding RNA through RGG box. EMBO J 1992;11:2655-64.
53. Ouyang Y, Salstrom J, Diaz-Perez S, Nahas S, Matsuno Y, Dawson D, Teitell MA, Horvath S, Riggs AD, Gatti RA, Marahrens Y. Inhibition of Atm and/or Atr disrupts gene silencing on the inactive X chromosome. Biochem Biophys Res Commun 2005;337:875-80.
54. Ariyoshi M, Schwabe JW. A conserved structural motif reveals the essential transcriptional repression function of spen proteins and their role in developmental signaling. Genes Dev 2003;17:1909-20.
55. Colley SM, Iyer KR, Leedman PJ. The RNA coregulator SRA, its binding proteins and nuclear receptor signaling activity. IUBMB Life 2008;60:159-64.
56. Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR. M6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016;537:369-73.
57. Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, Hamakubo T. Identification of Wilms' tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem 2013;288:33292-302.
58. Hasegawa Y, Brockdorff N, Kawano S, Tsutui K, Tsutui K, Nakagawa S. The matrix protein hnRNP U is required for chromosomal localization of xist RNA. Dev Cell 2010;19:469-76.
59. Tang L, Nogales E, Ciferri C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog Biophys Mol Biol 2010;102:122-8.
60. Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012;485:381-5.
61. Simmler MC, Cunningham DB, Clerc P, Vermat T, Caudron B, Cruaud C, Pawlak A, Szpirer C, Weissenbach J, Claverie JM, Avner P. A 94 kb genomic sequence 3' to the murine Xist gene reveals an AT rich region containing a new testis specific gene Tsx. Hum Mol Genet 1996;5:1713-26.
62. Ogawa Y, Lee JT. Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol Cell 2003;11:731-43.
63. Chureau C, Prissette M, Bourdet A, Barbe V, Cattolico L, Jones L, Eggen A, Avner P, Duret L. Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res 2002;12:894-908.
64. Augui S, Filion GJ, Huart S, Nora E, Guggiari M, Maresca M, Stewart AF, Heard E. Sensing X chromosome pairs before X inactivation via a novel X-pairing region of the Xic. Science 2007;318:1632-6.
65. van Bemmel JG, Mira-Bontenbal H, Gribnau J. Cis- and trans-regulation in X inactivation. Chromosoma 2016;125:41-50.
66. Zhang L, Huang H, Zhou F, Schimmel J, Pardo CG, Zhang T, Barakat TS, Sheppard KA, Mickanin C, Porter JA, Vertegaal AC, van Dam H, Gribnau J, Lu CX, ten Dijke P. RNF12 controls embryonic stem cell fate and morphogenesis in zebrafish embryos by targeting Smad7 for degradation. Mol Cell 2012;46:650-61.
67. Figueroa DM, Darrow EM, Chadwick BP. Two novel DXZ4-associated long noncoding RNAs show developmental changes in expression coincident with heterochromatin formation at the human (Homo sapiens) macrosatellite repeat. Chromosom Res 2015;23:733-52.
68. Deng X, Ma W, Ramani V, Hill A, Yang F, Ay F, Berletch JB, Blau CA, Shendure J, Duan Z, Noble WS, Disteche CM. Bipartite structure of the inactive mouse X chromosome. Genome Biol 2015;16:152.
69. Horakova AH, Moseley SC, Mclaughlin CR, Tremblay DC, Chadwick BP. The macrosatellite DXZ4 mediates CTCF-dependent long-range intrachromosomal interactions on the human inactive X chromosome. Hum Mol Genet 2012;21:4367-77.
70. Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, Huang SC, Sanborn AL, Machol I, Shamim M, Seberg AP, Lander ES, Chadwick BP, Aiden EL. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proc Natl Acad Sci U S A 2016;113:E4504-12.
71. Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, Dekker J. Structural organization of the inactive X chromosome in the mouse. Nature 2016;535:575-9.
72. Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, Guo N, Muruganujan A, Doremieux O, Campbell MJ, Kitano H, Thomas PD. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res 2005;33:D284-8.
73. Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 2017;45:D183-9.
74. Alvarez-Saavedra M, Sáez MA, Kang D, Zoghbi HY, Young JI. Cell-specific expression of wild-type MeCP2 in mouse models of Rett syndrome yields insight about pathogenesis. Hum Mol Genet 2007;16:2315-25.
75. Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, Foust KD, Kaspar BK, Bird A, Mandel G. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci 2013;33:13612-20.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Przanowski* P, Waśko* U, Bhatnagar S. Novel molecular players of X chromosome inactivation: new technologies and new insights. J Transl Genet Genom 2018;2:2. http://dx.doi.org/10.20517/jtgg.2017.03
AMA Style
Przanowski* P, Waśko* U, Bhatnagar S. Novel molecular players of X chromosome inactivation: new technologies and new insights. Journal of Translational Genetics and Genomics. 2018; 2: 2. http://dx.doi.org/10.20517/jtgg.2017.03
Chicago/Turabian Style
Przanowski*, Piotr, Urszula Waśko*, Sanchita Bhatnagar. 2018. "Novel molecular players of X chromosome inactivation: new technologies and new insights" Journal of Translational Genetics and Genomics. 2: 2. http://dx.doi.org/10.20517/jtgg.2017.03
ACS Style
Przanowski*, P.; Waśko* U.; Bhatnagar S. Novel molecular players of X chromosome inactivation: new technologies and new insights. J. Transl. Genet. Genom. 2018, 2, 2. http://dx.doi.org/10.20517/jtgg.2017.03
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.