
Reversal of X chromosome inactivation: lessons from pluripotent reprogramming of mouse and human somatic cells


Abstract
X chromosome inactivation (XCI) is a strategy used by mammals to silence genes along one of the two female X chromosomes and equilibrate expression dosage between XY males and XX females. This epigenetically-inherited silencing is established during early embryonic development and maintained thereafter through cell divisions. Seeding of multiple repressive epigenetic marks along the inactive X chromosome (Xi) makes inactivation extremely robust and difficult to reverse upon single genetic perturbations. Reversal of XCI has, however, been observed when somatic cells are reprogrammed towards pluripotency, and in vitro reprogramming techniques have been used in recent years to dissect Xi gene reactivation mechanisms. These studies pave the way for developing novel therapeutic approaches for X-linked diseases. Here, the author reviews Xi reactivation during pluripotent reprogramming of mouse and human somatic cells, highlight recent advances and species-specific differences, and discuss the relevance for human diseases.
Keywords
Introduction
Dosage compensation between XX females and XY males is achieved in placental mammals by the random inactivation of one of the two female X chromosomes[1,2]. This process - named X chromosome inactivation (XCI) - is a major example of epigenetic gene regulation that leads to global chromatin condensation and transcriptional silencing along the future inactive X chromosome (Xi)[2]. The choice of which X chromosome to inactivate is completely random and, once established in early embryonic precursors it is stably maintained through cell divisions. As a consequence, females are mosaics of cells expressing either the maternal or paternal allele that co-exist in a 50:50 ratio. Mosaicism makes females more refractory to diseases that are caused by X-linked mutations because cells expressing the wild-type allele can functionally compensate the deficit in heterozygous carriers. In addition, the X chromosome that carries mutations or structural abnormalities is often found as inactive in the majority of somatic cells due to selection[3,4]. Many X-linked diseases are indeed asymptomatic in heterozygous females and manifest only in their male progeny[5]. However, some X-linked mutations are lethal in males and lead to severe disease in heterozygous females[6]. For example, in the case of gene mutations with a dominant negative effect, expression of a wild-type allele from 50% of cells is not able to rescue the function in heterozygosity. This is exemplified by UBQLN2 mutations that cause abnormal protein aggregation and consequent neurodegeneration in X-linked amyotrophic lateral sclerosis[7]. Importantly, there are other diseases in which the compensatory effect of XCI in heterozygous females is not effective due to tissue-specific sensitivity to dosage unbalance and/or to skewed XCI patterns that favor the expression of mutated genes. For example, expression of the wild-type allele only from half of the cells may lead to loss of function only in certain tissues but not in others, as for mutations in the X-linked MECP2 gene that specifically cause a severe brain deficit even if the gene is constitutively expressed[8,9]. In addition, in human some genes escape inactivation and are variably expressed from the Xi in different tissues of the same individual and even in-between different females[10]. This variability in Xi expression has been suggested to influence the penetrance and expressivity of X-linked diseases as hypothesized for oro-facial-digital syndrome type 1[11]. Finally, it has been shown that XCI skewing can also favor cells in which the wild-type allele is on the inactive X chromosome thus leading to the outbreak of disease in heterozygous females[12-16].
An important implication of the epigenetic regulation of Xi gene expression is the preservation of the genetic material and, indeed, the presence of a silent set of alleles for over a thousand genes on the X chromosome. Reactivation of wild-type alleles in heterozygous females could potentially be harnessed and might represent a therapeutic approach for many X-linked diseases. This is probably best exemplified by Rett syndrome, for which a phenotypic reversal of advanced neurological symptoms has been shown in both young and adult mice upon expression of a wild-type MeCP2[17]. Importantly, rescue of disease symptoms has also been observed in human and mouse upon gene therapy of X-linked adrenoleukodystrophy[18] and Hunter syndrome[19,20], two recessive X-linked diseases that also affect heterozygous females[16,21]. This suggests that the usefulness of Xi reactivation in clinical approaches might be extended beyond dominant X-linked diseases. Understanding the molecular mechanisms of Xi inactivation and reactivation will be instrumental in the future to engineer selective Xi gene reactivation as a novel clinical approach for several X-linked diseases.
Our current understanding of XCI and its molecular mechanisms derives from studies in the mouse. During mouse embryonic development, Xi gene silencing is triggered by Xist, a long non-coding RNA that is up-regulated from the future Xi and spreads along the chromosome domain[22]. Although the repertoire of Xist interacting proteins has been recently identified, the precise molecular mechanisms by which Xist initiates silencing are still unknown[23-27]. It is currently believed that Xist might act as a scaffold to recruit further factors on the Xi, including Polycomb repressive complex 1 (PRC1), histone deacetylases, histone variants and the DNA methylation machinery[24,25,28-30]. Upon Xist coating the inactive X chromosome indeed becomes progressively devoid of RNA polymerase II and chromatin marks associated with transcriptionally active regions[31]; whereas it is enriched of repressive marks such as H3K27me3[32], H3K9me2/me3[33], H2AK119ub[34,35], macroH2A1[36] and promoter DNA methylation[37]. The multitude of epigenetic modifications that are deposited along the Xi are believed to maintain silencing in a redundant manner as removing single factors does not lead to global gene reactivation. Supporting this hypothesis, it has been shown that Xi gene silencing is maintained in the absence of macroH2A1 histone variant[38] and upon functional loss of Polycomb repressive complex PRC2 and some PRC1 components, which catalyze the deposition of the repressive chromatin marks H3K27me3 and H2AK119ub respectively[39,40]. A recent study of several Xist interacting proteins has also shown that knock-down of any single interactor could not reactivate an Xi-integrated GFP reporter, whereas combining targeting of each interactor with inhibition of DNA methylation and topoisomerases leads to reactivation of 75-100 Xi genes out of around 200 analyzed[25]. On the other hand, depletion of Xist in somatic cells leads to a stochastic reactivation of single Xi genes[41] that can be potentiated when combined with histone deacetylase (HDAC) inhibitors and DNA demethylation[42]. Importantly, Xist depletion in mouse fibroblasts has been shown to alter the Xi chromosome conformation making it more similar to the one of the active X chromosome (Xa)[25]. In addition, silencing of factors that affect Xist expression and/or localization has been shown to partially reactivate the Xi[43,44]. Altogether these findings support the hypothesis that multiple epigenetic layers are in place to prevent Xi reactivation and suggest a central role for Xist both in the initiation and maintenance of XCI.
Cell fate reprogramming studies offer the opportunity to model Xi reactivation and unravel its molecular mechanisms. Reversal of X inactivation has, in fact, been observed when mouse somatic cells are reprogrammed towards a pluripotent state. A tight link between pluripotency and reversal of XCI has been demonstrated[45-48], and reprogramming of somatic cells towards pluripotency has been widely used to investigate its dynamics and molecular mechanisms in mouse[49-52]. In human, the connection between pluripotency and Xi reactivation has instead been controversial as different Xi states have been reported in human embryonic stem cells (ESCs)[53-57] and in induced pluripotent stem cells (iPSCs)[55-59]. The lack of defined culture conditions that stabilize an embryonic-like pluripotent state and the resulting epigenetic instability of human pluripotent cells contributed to the observed variability. Recent studies in human embryos and ESCs suggest substantial differences in the mechanisms of XCI between human and mouse thus highlighting the need for model systems that allow to directly investigate the human Xi and its reactivation[57,60-62]. Here, I review studies of Xi reactivation during pluripotent reprogramming of both mouse and human cells, emphasize specie-specific differences and recent advances in reprogramming-mediated human Xi reactivation.
Reprogramming-mediated Xi reactivation in mouse
In mouse embryos, the two X chromosomes undergo several rounds of inactivation and reactivation[63,64][Figure 1A and B]. The first round of XCI takes place at the 4-cell stage and leads to the inactivation of the paternally-inherited X chromosome. This imprinted form of XCI is followed by reactivation in the epiblast cells of the blastocyst where random XCI is later established around implantation. Imprinted XCI is instead maintained in extraembryonic lineages. Interestingly, reversal of X inactivation has also been observed during mouse development when pluripotency genes are re-expressed in primordial germ cells (PGCs)[65-67]. In addition, several mouse pluripotency factors have been shown to inhibit XCI[45-48] thus suggesting an intimate connection between pluripotency and the presence of two active X chromosomes.
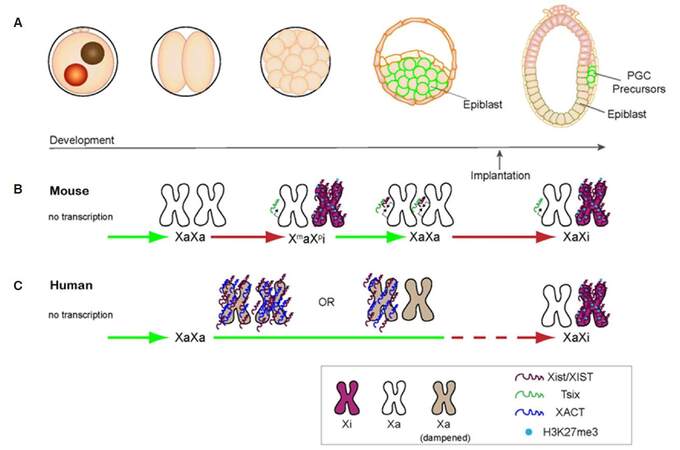
Figure 1. X chromosome state in mouse and human female embryos. (A) Schematic of early developmental stages; (B) in mouse embryos, both X chromosomes are transcribed upon the zygote genome activation (ZGA) at 2-cell stage. Imprinted inactivation of the paternal X chromosome then takes place at the 4-cell stage and is followed by reactivation in the epiblast that will give rise to the embryo proper. Upon implantation, the epiblast undergoes random XCI whereas imprinted XCI is maintained in extraembryonic lineages. As depicted, mouse Xi is characterized by the expression and coating of Xist, whereas the Xa expresses Tsix, a long non-coding RNA that antagonizes Xist and functions specifically in mouse but not in human; (C) in human embryos, the two X chromosomes are transcribed upon ZGA at 4-cell stage and remain transcriptionally active throughout pre-implantation development. XIST and another human-specific long non-coding RNA, XACT, coat both active X chromosomes in human embryos. Notably, XIST RNA does not tightly localize on the X chromosomes but shows a diffuse pattern and its coating is not accompanied by enrichment of H3K27me3. H3K27me3 enrichment is instead observed along the Xi upon XCI initiation. XCI: X chromosome inactivation; Xi: inactive X chromosome; Xa: active X chromosome; PGC: primordial germ cell
Cell fusion-mediated Xi reactivation
The earliest evidence of an association between reversal of XCI and pluripotent reprogramming of somatic cells is dated back to 1983 when Takagi et al.[68] showed that fusions between female mouse thymocytes and pluripotent mouse embryonic carcinoma cells (ECCs) shifted the replication timing of the thymocyte Xi from late to early S phase, as observed for transcriptionally active X chromosomes. Early replication timing was associated with reactivation of the X-linked Pgk1 gene from the Xi and was observed upon fusions of several differentiated cells from thymus, spleen or bone marrow with ECCs, but not when two differentiated cell types were fused together. Importantly, hybrids obtained between mouse somatic cells and ECCs acquired the pluripotent differentiation potential of the parental carcinoma cells suggesting that reprogramming towards pluripotency might trigger Xi reactivation. Later studies confirmed that embryonic pluripotent cells of different origin (including ECC, ESC and embryonic germ cells, EGC) are able to reprogram the somatic cell genome and reverse XCI upon fusion. Specifically, it has been shown that the somatic Xi acquires epigenetic features of the active X chromosome (Xa), including early replication timing, loss of Xist coating and Xi-associated histone marks (e.g., H3K27me3 and H2Aub1), and re-expresses an Xi-linked GFP transgene[69-71]. Importantly, somatic-ESC hybrids are able to re-initiate random XCI upon differentiation suggesting a complete erasure of epigenetic memory along the Xi[70,72]. However, the fusion of ESC with somatic cells does not lead to erasure of genomic imprinting[69,73]. This extended reprogramming potential has instead been observed in fusions with EGCs that can induce both Xi reactivation and loss of DNA-methylation imprinting marks in the somatic nuclei[73,74].
Cell fusion studies have also demonstrated that the reprogramming capacity of embryonic pluripotent cells is ascribed to the transfer of pluripotency-associated transcriptional factors into the somatic nucleus[71,75]. Specifically, it has been shown that mouse ESCs (mESCs) overexpressing Nanog have an enhanced reprogramming capacity whereas ESCs lacking Oct4, but not Sox2, fail to induce pluripotent reprogramming in the somatic fusion partner. This is consistent with findings in mESCs where Oct4, Nanog, Sox2 and Rex1 repress Xist expression, while Klf4, c-Myc and Rex1 up-regulate Tsix[45-48], a long noncoding RNA that antagonizes Xist and protects the Xa from inactivation[76-78]. These results suggest that pluripotency factors within the mESCs might induce Xi reactivation by directly repressing Xist and activating Tsix. Consistently with this hypothesis, re-expression of Oct4 from the somatic genome has been shown to precede Xi reactivation and repression of Xist expression upon cell fusion-mediated reprogramming[50]. The delayed repression of Xist has been associated with the kinetics of DNA methylation at its 5’ regulatory sequences and is enhanced by the activation of Dnmt3a and Tsix via HDAC inhibitors. This suggests that chromatin remodeling is required together with pluripotency factors in order to reverse XCI, although delocalization of Xist rather that loss of its expression has been associated with Xi gene reactivation[79].
Somatic cell nuclear transfer-mediated Xi reactivation
The somatic cell genome can restart the entire developmental program upon nuclear transfer into eggs or oocytes. Conversely to other reprogramming techniques, somatic cell nuclear transfer (SCNT) reprograms differentiated cells to a totipotent state from which both embryonic and extra-embryonic lineages develop into cloned animals[80,81]. In this model system, reprogramming is triggered by maternally-inherited factors that are present in the cytoplasm of metaphase II oocytes. Interestingly, it has been shown that the zygote and blastomeres of early embryonic stages are not capable of reprogramming somatic nuclei unless they are in mitosis[82,83]. The enhanced reprogramming capacity of mitotic cells has been ascribed to the release of chromatin-bound factors during mitosis and depends upon cell cycle synchronization between the somatic donor and the embryonic recipient[84,85]. This evidence suggested a model in which the mitotic environment of the recipient cell induces premature chromatin condensation and consequent release of chromatin-bound factors in somatic nuclei, thus facilitating genomic access of transcriptional regulators that re-establish a totipotent transcriptional program[86].
The success of nuclear reprogramming has been associated with erasure of epigenetic memory from the somatic nucleus[87] and the reactivation of the somatic Xi has been used as a model to investigate epigenetic reprogramming. Initial experiments by Eggan et al.[49] showed that the reactivation of a GFP transgene on the somatic Xi occurs by the morula/blastocyst stage and is followed by random XCI in the embryonic lineages. In the extraembryonic cells, instead, the somatic Xi is re-inactivated suggesting an incomplete erasure of epigenetic memory in this lineage, in which imprinted inactivation of the paternal X chromosome occurs during normal development. Consistently with this hypothesis, random XCI was observed in the extraembryonic lineages when mESCs were used as donors for nuclear transfer. This most likely results from the failure in resetting imprinting marks of donor nuclei[88], which are instead erased during normal development in primordial germ cells and re-established in the gametes in a parent-specific manner[89]. Further studies confirmed that cloned embryos fail to establish imprinted XCI in preimplantation stages and extraembryonic tissues, and revealed heterogeneity in random XCI within cloned embryos in which some cells undergo XCI while others do not inactivate any X chromosome[90]. Although progressive loss of Xist coating has been observed within 30 min after SCNT, the precocious appearance of H3K27me3 and H3K9me2 on the original Xi suggests an incomplete reprogramming of the somatic nucleus[91]. The extent of Xi gene reactivation remains however unknown. Altogether these studies suggest that nuclear transfer cannot fully erase Xi epigenetic marks during pre-implantation development and further reprogramming events are required for re-establishing the normal developmental program. Interestingly, ectopic Xist accumulation has been observed upon SCNT in both male and female embryos and loss or depletion of Xist have been associated with increased efficiency of reproductive cloning[92,93]. In order to get some mechanistic insights, it would be important to determine to which extent genes along the Xi are reactivated in single blastomeres and their correlation with the observed Xi epigenetic changes.
Xi reactivation has also been studied by injecting somatic nuclei into the germinal vesicle of Xenopus oocytes[51]. This study showed that the developmental state of the donor influences the reactivation of an X-linked transgene located on the Xi. Specifically, reactivation was observed when mouse post-implantation epiblast stem cells (EpiSCs) were injected into frog oocytes but not upon transfer of embryonic fibroblasts or extraembryonic cell nuclei. Comparative Xi chromatin analysis showed loss of Xi-associated Xist both in EpiSCs and reactivation-resistant cells, whereas H3K27me3 and DNA methylation were invariably maintained. Accumulation of the histone variant macroH2A was instead observed on the Xi of reactivation-resistant cells but not in EpiSCs. MacroH2A depletion upon transfer of fibroblast nuclei lead to partial Xi reactivation that could be enhanced when it was combined with HDAC inhibitors or activation of Oct4 and Sox2. This suggests that macroH2A contributes to the stability of Xi but other factors are required for full Xi reactivation. Notably, it has been shown that in human somatic cells macroH2A association with the Xi is cell-cycle dependent being most prominent in early S phase and declining from late S through mitosis[94]. Nonetheless, macroH2A is retained on the Xi during mitosis whereas human XIST and other Xi-associated histone marks (e.g., H2A ubiquitination) are lost[95]. As cell cycle has been shown to influence the efficiency of SCNT-reprogramming (discussed above) and mouse Xist is instead associated with the Xi throughout mitosis, it will be interesting to determine whether the developmental state of the donor nucleus and/or its cell cycle phase also influence Xi reactivation and investigate mechanistic differences between mouse and human.
iPSC-mediated Xi reactivation
Global epigenetic resetting has been observed upon induction of pluripotency by transduction of four pluripotency factors (i.e., Oct4, Sox2, Klf4 and c-Myc) into mouse fibroblasts[96,97]. Similarly to mESCs, miPSCs have two active X chromosomes and undergo de novo random XCI upon differentiation[97]. Recent studies have indeed used iPSC reprogramming to dissect the molecular mechanisms of Xi reactivation in mouse[52,98].
A tight association between the reversal of XCI and the sequential activation of pluripotency factors has been detected by investigating the kinetics of Xi epigenetic changes during mouse iPSC reprogramming[52]. Specifically, it has been shown that loss of Xist from the Xi follows Nanog expression, consistently with Nanog role in repressing Xist expression. Xi gene reactivation was instead observed in a subset of Nanog positive cells that reactivate additional factors (i.e., DPPA4 and PECAM1) at later reprogramming stages. This suggests that the hierarchical activation of pluripotency factors is required for complete reversal of XCI[52,99]. Consistently with this hypothesis, depletion of Nanog impaired Xi reactivation, whereas its overexpression during late reprogramming stages promoted the formation of iPSC colonies expressing DPPA4 and bi-allelically transcribing Tsix from both X chromosomes. These data suggest that Nanog expression is required but not sufficient for efficient reversal of XCI. A further link between pluripotency and Xi reactivation has been provided by Prdm14, a germline factor that has been implicated in the epigenetic reprogramming of PGCs and whose expression correlates with Xi reactivation[100]. It has been shown that depletion of Prdm14 during iPSC reprogramming decreases Xi reactivation and hampers both the derivation and maintenance of iPSC colonies[98]. Prdm14 overexpression in mouse EpiSCs instead induced efficient conversion to ESCs and Xi reactivation[101]. Mechanistically, Prdm14 has been shown to repress Xist in a dual manner. First, it represses Rnf12, a E3-ubiquitin ligase that targets Rex1 for proteosome degradation during differentiation[48]. Second, it directly binds Xist in a Tsix-dependent manner. Interestingly, depletion of Tsix during iPSC reprogramming does not compromise Xi reactivation neither iPSC colony formation[52,98], although it affects Xist repression and Xi reactivation in the mouse embryonic epiblast[102].
iPSC reprogramming studies also gave further mechanistic insights in the role of Xist during Xi reactivation[52]. Kinetic studies during iPSC reprogramming showed that loss of Xist RNA in Nanog positive cells precedes bi-allelic expression of X-linked genes, suggesting that Xist repression might be required for Xi gene reactivation. Supporting this hypothesis, constitutive Xist expression decreased the reactivation of Xi genes within Nanog positive cells without affecting reprogramming, whereas Xist depletion did not alter Xi reactivation events neither its kinetics. This suggests that Xist repression might be necessary but insufficient for Xi gene reactivation. Notably, bi-allelic expression of X-linked genes was enhanced upon combined deletion of Xist and inhibition of DNA methylation, thus supporting the hypothesis that Xi reactivation requires both Xist silencing and DNA demethylation.
Reprogramming-mediated Xi reactivation in human
Recent studies in human embryos have shown substantial differences between human and mouse XCI[60,61,103][Figure 1A and C]. Differences concern both the status of the female X chromosomes in pre-implantation embryos and the factors involved in the regulation of XCI. It has been shown that the two female X chromosomes are transcriptionally active throughout human pre-implantation development and both in embryonic and extraembryonic lineages[60]. This contrasts with mouse embryos where several rounds of Xi reactivation/inactivation take place in the epiblast cells that give rise to the embryo proper [Figure 1B], whereas imprinted XCI of the paternally-inherited chromosome is maintained in extraembryonic cells[104]. Random XCI initiates in mouse epiblast cells around the time of implantation and is essential for proper development[63,64,105,106], while its precise timing is unknown in human[60]. Furthermore, a recent single cell RNA-seq study showed a progressive downregulation of both X chromosomes beyond the zygote genome activation stage through human preimplantation development, suggesting that a different dosage compensation mechanisms might be in place ahead of XCI[61]. Consistently with this hypothesis, it has been observed that human XIST RNA coats both X chromosome by the morula and blastocyst stages[60,61,103]. However, whether bi-allelic accumulation of XIST leads to dampening of X-linked gene expression remains to be determined. Interestingly, a human-specific long non-coding RNA, named XACT, has been shown to localize to either one or both X chromosomes in human embryos and has been hypothesized to compete with XIST for binding along the chromosome[103,107]. Notably, XIST RNA signal has a dispersed nuclear localization in human embryos suggesting that it might be delocalized from Xi chromatin or bind only some chromosomal domains[60,103].
Xi reactivation in human pluripotent cells
Human pluripotent stem cells (i.e., hESCs and hiPSCs) are epigenetically unstable ex vivo and heterogeneous in their XCI state that varies between different cell lines and also among cells of the same culture[53-59,108,109]. Three different classes of hESCs have been originally described based on XIST nuclear pattern and the transcription of CotI repeats before and after differentiation[54]. Class I hESCs were characterized by the absence of XIST RNA signal and the presence of CotI transcripts from both X chromosomes. Upon differentiation of these hESCs, XIST was upregulated and coated one of the two X chromosomes whereas CotI transcription within the XIST-coated domain ceased. Collectively these results suggested that class I hESC retain two transcriptionally active X chromosomes and are able re-initiate XCI. XIST-coating and CotI exclusion, however, were shown to occur spontaneously when class I cells were maintained in culture, thus generating XaXi XIST+ hESCs (class II). Finally, class III hESCs were observed upon progressive loss of XIST-coating in class II cultures. Notably, loss of XIST nuclear domain was associated with promoter DNA demethylation and transcriptional reactivation of some Xi genes[53,56]. However, these class III hESCs could not re-establish XIST expression neither Xi gene silencing upon differentiation and were considered subject to an “erosion” of dosage compensation[53,54,59,110]. Interestingly, Xi reactivation occurring during erosion was shown to localize preferentially within chromosomal domains enriched with H3K27me3[107]. These H3K27me3 domains are lost upon erosion whereas H3K9me3-enriched domains, which spatially segregate along the human Xi, remain unaffected. Surprisingly, loss of XIST is not directly associated with Xi gene reactivation as it occurs afterwards. Reactivation and accumulation of XACT RNA along the Xi was instead observed ahead of XIST loss and Xi gene reactivation [Figure 2A] and was indeed hypothesized to trigger the reorganization of Xi heterochromatin domains and consequent gene reactivation. As XACT coats the active X chromosome in XaXi class II hESCs and in the human epiblast ahead of XCI, it has been suggested to antagonize XIST by competing for Xi binding or limiting chromosome accessibility[103,109]. Consistently with this hypothesis, overexpression of a XACT transgene in mESCs has been shown to prevent inactivation of the X chromosome from which it is expressed, whereas its downregulation restores random XCI[103].
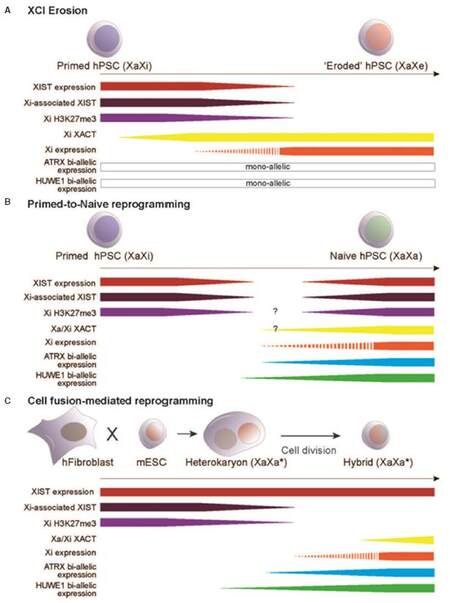
Figure 2. Human Xi reactivation in pluripotent cells. Schematic representation of epigenetic changes that are associated with Xi reactivation in different model systems. (A) Erosion of XCI in primed human pluripotent cells (i.e., ESCs and iPSCs) leads to partial Xi gene reactivation that leads to the "eroded" X chromosome (Xe). Sequential Xi chromatin changes occur ahead of gene reactivation and include XACT re-expression and Xi localization followed by loss of Xi-associated XIST and H3K27me3[107]. The extent of Xi reactivation and the reactivated genes vary in different cell lines. Xi genes that are refractory to erosion (i.e., HUWE1 and ATRX) remain mono-allelically expressed; (B) reprogramming of primed human pluripotent cells to the naïve state is represented accordingly to 5iLAF[116]. XIST expression and its enrichment on the human Xi (i.e., Xi-associated XIST) are transiently lost during the transition from the primed (XaXi XIST-expressing) to naïve (XaXa XIST-expressing) state[62]. No data is available for H3K27me3 enrichment on the Xi neither for XACT relative to the timing of transient XIST loss (highlighted by question marks); (C) reprogramming of human somatic cells by fusion with mouse ESCs induces partial Xi gene reactivation (Xa*) ahead of cell division[126]. In this system, loss of H3K27me3 enrichment and XIST delocalization from the Xi are early chromatin changes that precede Xi gene reactivation. Importantly Xi genes that are refractory to erosion show bi-allelic expression in the subset of cells where XIST is delocalized. XACT re-expression is a later event and is not associated with Xi gene reactivation. XCI: X chromosome inactivation; Xi: inactive X chromosome; Xa: active X chromosome; Xp: paternal X chromosome; PSC: pluripotent stem cell; ESC: embryonic stem cell
The variable XCI state and its progressive epigenetic alterations in hESCs have been suggested to result from inappropriate culture conditions that are unable to stabilize the XCI state during the derivation from the in vivo epiblast and further in vitro passaging[111]. Derivation in low oxygen levels allowed to obtain hESCs with two active X chromosomes and was suggested to preserve this state[112]. However, a separate study showed that hypoxia rather stabilize hESCs that have already undergone XCI[113]. Other studies reported the derivation of hESCs that preserved the ground state of the in vivo epiblast but they did not fully characterize the status of the X chromosomes[114-116]. In light of recent findings showing that the two X chromosomes in human female embryos are characterized by dual XIST coating and dampening of X chromosome expression, a recent work highlights the importance of characterizing chromosome-wide X-linked gene expression and XIST nuclear localization before and after differentiation[57]. Multi-gene RNA-FISH and allele-specific X-linked gene expression revealed that hESC derived and propagated in standard FGF2-containing medium maintain their XCI state upon differentiation. Notably, it has been shown that XIST-XaXa hESCs (previously defined as class I) cannot re-express XIST neither undergo XCI upon differentiation similarly to cells in the XaXe eroded state. These aberrant cells arise from blastocyst outgrowth as early as 48 h after plating onto feeders and can be stably maintained in this state upon establishment and propagation of hESCs. Although it cannot be formally excluded that these cells represent an intermediate state in human XCI, they are currently believed to result from epigenetic adaptation to in vitro culture.
Similar epigenetic instability has been shown to occur in human iPSCs and it is probably the cause of controversial results in different labs[58,59,117-121]. Some groups, in fact, reported that XCI is maintained upon human iPSC reprogramming[58,59,117,119] while others claimed Xi reactivation[55,118,120,121]. Most of these studies, however, analyzed indirect markers of Xi reactivation, such as XIST and H3K27me3 nuclear localization and expression of X-linked genes compared to autosomes, or directly assessed allele-specific expression of only few Xi genes. These analyses could easily confuse Xi reactivation with erosion, as it has been recently shown for hiPSC that were initially supposed to reactivate the Xi upon culture on LIF producing feeders. The latter have instead been shown to undergo extensive erosion of XCI by multi-gene RNA-FISH[57].
Recently, two culture conditions[116,122] have been shown to reprogram hESCs and hiPSCs to a state similar to human blastocysts[123]. Detailed analysis of XCI state in these “naive” human pluripotent cells showed that they retain some features of the epiblast, including dual XIST and XACT coating, bi-allelic expression of X-linked genes and dampening of X-linked gene expression on both X chromosomes[62,103][Figure 2B]. However, the same Xi was re-inactivated upon differentiation and only a minority of cells within the culture (< 10%) showed XIST-coating on both X chromosomes. Collectively, these results suggest that an improvement of culture conditions is still required for stabilizing human naïve pluripotency and careful X chromosome-wide analysis of Xi-specific expression need to be performed in the future to unequivocally define XCI state in pluripotent cells.
Xi reactivation by interspecies cell fusion-mediated reprogramming
Cell fusion between somatic and ESCs from different species has been used to investigate human pluripotent reprogramming[75,124]. This system allows the analysis of early reprogramming events because specie-specific features in nuclear organization (e.g., the presence of chromo centers in mouse nuclei) and DNA sequence differences can be used to track each fusion partner by imaging and molecular techniques. Importantly, cell nuclei remain separated within a shared cytoplasm and this transient heterokaryon state persists until the first mitosis, when nuclei fuse and generate hybrids[125]. We have, indeed, recently used cell fusion between human female fibroblasts and mouse ESCs to reprogram the somatic nucleus and investigate human Xi reactivation[126][Figure 2C]. We showed that expression of pluripotency genes from the human nucleus occurs as early as two days after fusion at a time when the majority of cells are heterokaryons. This observation allowed us to discriminate pre- and post-mitotic reprogramming events in heterokaryons and hybrids, respectively. Single cell analyses demonstrated that XIST delocalization and loss of H3K27me3-enrichment from the human Xi occur in heterokaryons and hybrids 2-3 days after fusion, and precede bi-allelic expression of ATRX and HUWE1, two X-linked genes that are not subject to XCI erosion in human ESCs[57,107]. RNA-FISH analysis of nascent ATRX and HUWE1 transcripts together with XIST or XACT RNAs showed that bi-allelic expression of X-linked genes only occurs in cells that have lost a localized XIST signal (i.e., about 30% at 3 days after fusion) but not in all of them (50% at day 3). XACT instead re-associates with either one or two X chromosomes later during reprogramming in a minority of both mono-allelic and bi-allelic cells (< 1% at day 6). These results suggest that human Xi reactivation is induced by cell fusion-mediated reprogramming ahead, or immediately after, cell division and that it requires XIST delocalization, but not XACT re-association along the Xi. Notably, delocalization of XIST upon cell fusion-mediated reprogramming results in a diffuse nuclear signal that resembles the one observed in human blastocysts and might represent remaining binding at foci from where XIST initially spreads[127,128]. In addition, a minority of heterokaryon and hybrid nuclei (7% at day 3 vs. 1% at day 0; unpublished data) have two diffused XIST clouds suggesting that cell fusion-mediated reprogramming might be able to recapitulate some features of the in vivo epiblast cells. Consistently with this hypothesis, genome-wide expression analysis upon cell fusion-mediated reprogramming showed the reactivation of pluripotency genes associated with both primed and naïve human pluripotent cells[129]. In the future, single cell studies might indeed be used to segregate different human pluripotent states and help us refine culture conditions for naïve pluripotency.
To investigate the extent of Xi gene reactivation along the entire X chromosome, we derived human fibroblast clones with reciprocal Xi chromosomes (e.g., Xa1Xi2 and Xi1Xa2) and performed allele-specific RNA-seq analysis of these clones before and after reprogramming[129]. This analysis showed that cell fusion-mediated reprogramming induces partial human Xi reactivation with 10% of the sampled genes being consistently reactivated at different times after fusion and in different clones. Notably, clones with opposite Xi haplotype reactivate the same set of genes suggesting that neither parent of origin nor mutations in regulatory sequences influence reactivation. We instead observed a preferential reactivation of Xi genes localized within XCI “escape” domains at the telomeric end of the short Xp arm suggesting a higher susceptibility of these loci to reactivation. Similar results were obtained in a large iPSC population study that identified Xi domains reactivating with higher frequency[130]. Conversely to iPSC erosion, however, Xi genes that were reactivated upon cell fusion-mediated reprogramming could not be predicted by H3K27me3 domains suggesting that the mechanism leading to reactivation might be different in these two systems[107]. Cell fusion-mediated Xi reactivation could instead be predicted by variable Xi expression among single cell-derived fibroblast clones ahead of reprogramming. This probably reflects an intrinsic predisposition of certain loci to transcriptional activation that can already occur in the somatic cells with low frequency and is enhanced upon reprogramming. Interestingly, recent studies of three-dimensional X chromosome organization in mouse suggested that genes variably expressed from the Xi in a cell-specific context co-localize within topologically associated domains and showed reduced expression upon disruption of the domain architecture[131]. As loci that escape X inactivation vary between different cell types and also between females[10], future population analysis of somatic cells from different tissues will help us to understand the molecular basis for this higher reactivation susceptibility.
Another important finding of our Xi reactivation study is the possibility of using cell fusion-mediated reprogramming to identify distinct subsets of Xi genes based on their susceptibility to reactivation upon further epigenetic perturbations. Indeed, DNA demethylation of human fibroblasts allowed us to identify a second subset of genes that was reactivated only after reprogramming. This suggests that the extensive Xi chromatin remodeling that takes place during reprogramming might unmask limiting factors in the reactivation of Xi gene subsets.
Final remarks
Pluripotent reprogramming of somatic cells has been widely used to induce the reversal of X chromosome inactivation and investigate its molecular mechanisms. In the last decade, mouse somatic cell reprogramming allowed major steps forward in unraveling the molecular connections between pluripotency and X chromosome inactivation/reactivation[51,52,71,98]. Reprogramming, indeed, represents a promising system to investigate the functional role of newly discovered XCI factors[23-27].
In light of recent findings in human embryos, the interpretation of mouse XCI studies need to be carefully considered when translated to human. Recent analyses of human pre-implantation development show remarkable differences between the events and the molecular players that characterize X chromosome inactivation in human and mouse, suggesting unforeseen specie-specific mechanisms[60,61,103][Table 1]. In addition, the epigenetic instability and heterogeneity of human pluripotent cells upon time in culture limit the possibility of modelling human XCI and its reversal in vitro. Recently, comparison of transcriptome profiles of single epiblast cells with human pluripotent cells allowed the identification of culture conditions that better preserve naïve pluripotency ex vivo[116,122,123]. XCI state, however, cannot be fully recapitulated in these cultures[62,103] thus highlighting the need for further improvements. We propose that cell fusion-mediated reprogramming might be used to segregate different pluripotent states with distinct X chromosome epigenetic features in single cells, as it can induce the expression of genes associated with both the primed and naïve human pluripotent states[129]. This information might be useful for identifying pathways that stabilize naïve pluripotency and for refining culture conditions.
Differences between mouse and human X chromosome inactivation
| Mouse | Human | |
|---|---|---|
| Random XCI initiation | Late blastocyst | Unknown |
| X chromosome dampening | Not detected | Dosage compensation mechanism (pre-implantation) |
| Imprinted XCI (embryonic) | Xp inactivation (4-cell stage) /reverted in epiblast (mid-blastocyst stage) | Not detected |
| Imprinted XCI (extra-embryonic) | Xp inactivation (mantained in trophoectoderm) | Not detected |
| XIST expression and localization | Mono-allelic Xi expression and coating upon random XCI | Bi-allelic expression and XaXa coating ahead of random XCI |
| Tsix/TSIX expression and function | Xa expression; Xist antagonist | No expression (pre-implantation)/ Xi expression (foetal cells); unknown function |
| XACT expression and function | No orthologue identified | XaXa expression and coating ahead of random XCI; XIST antagonist? |
The controversies regarding XCI state in human pluripotent cells also highlight the importance of standardized protocols for determining XCI status. Indirect measurements of XCI, such as the presence of XIST nuclear foci and/or the expression ratio between X and autosomes, as well as analyses of few X-linked genes with only one methodology (e.g., RNA-FISH or allele-specific RT-PCR) might be misleading[57]. The advent of genome-wide techniques that can be used to assess allele-specific expression at the single cell level will be fundamental in defining different XCI states and investigating the susceptibility of distinct Xi loci to reactivation. In addition, it will be important to reach an agreement about whether Xi expression should be defined as percentage of expression relative to the Xa or based on more sophisticated statistical methods[129,132-134]. Notably, the combination of molecular and cell biology techniques applied to human pluripotent cells before and after differentiation is required to distinguish naïve from primed and eroded pluripotency. Finally, another important aspect to consider is the variability of XCI in different tissues and individuals[10]. This variable Xi expression will need to be taken into account, and studies of XCI in a large number of subjects and in different tissues will be required to achieve a better understanding of how cellular context influence locus susceptibility to reactivation.
In perspective, studies of human pluripotent reprogramming will allow us to dissect the precise molecular mechanisms of Xi reactivation and to unravel locus-specific susceptibilities to reactivation. This information might help us to achieve locus-specific control of Xi reactivation and to engineer novel therapeutic strategies for X-linked diseases that will be based on the re-expression of the silent allele from the Xi. Furthermore, these studies will give us mechanistic insights into human diseases in which the reactivation of genes along the Xi has been observed, including cancer[135-137], age-related[138,139] and autoimmune diseases[140].
Declarations
Authors’ contributionsI. Cantone contributed solely to the paper.
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2017.
REFERENCES
2. Deng X, Berletch JB, Nguyen DK, Disteche CM. X chromosome regulation: diverse patterns in development, tissues and disease. Nat Rev Genet 2014;15:367-78.
3. Schmidt M, Du Sart D. Functional disomies of the X chromosome influence the cell selection and hence the X inactivation pattern in females with balanced X-autosome translocations: a review of 122 cases. Am J Med Genet 1992;42:161-9.
4. Peeters SB, Yang C, Brown CJ. Have humans lost control: the elusive X-controlling element. Semin Cell Dev Biol 2016;56:71-7.
5. Dobyns WB, Filauro A, Tomson BN, Chan AS, Ho AW, Ting NT, Oosterwijk JC, Ober C. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet A 2004;129A:136-43.
6. Franco B, Ballabio A. X-inactivation and human disease: X-linked dominant male-lethal disorders. Curr Opin Genet Dev 2006;16:254-9.
7. Daoud H, Rouleau GA. A role for ubiquilin 2 mutations in neurodegeneration. Nat Rev Neurol 2011;7:599-600.
8. Sugino K, Hempel CM, Okaty BW, Arnson HA, Kato S, Dani VS, Nelson SB. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J Neurosci 2014;34:12877-83.
9. Ross PD, Guy J, Selfridge J, Kamal B, Bahey N, Tanner KE, Gillingwater TH, Jones RA, Loughrey CM, McCarroll CS, Bailey ME, Bird A, Cobb S. Exclusive expression of MeCP2 in the nervous system distinguishes between brain and peripheral Rett syndrome-like phenotypes. Hum Mol Genet 2016;25:4389-404.
11. Morleo M, Franco B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J Med Genet 2008;45:401-8.
12. Badens C, Martini N, Courrier S, DesPortes V, Touraine R, Levy N, Edery P. ATRX syndrome in a girl with a heterozygous mutation in the ATRX Zn finger domain and a totally skewed X-inactivation pattern. Am J Med Genet A 2006;140:2212-5.
13. Bicocchi MP, Migeon BR, Pasino M, Lanza T, Bottini F, Boeri E, Molinari AC, Corsolini F, Morerio C, Acquila M. Familial nonrandom inactivation linked to the X inactivation centre in heterozygotes manifesting haemophilia A. Eur J Hum Genet 2005;13:635-40.
14. Viggiano E, Ergoli M, Picillo E, Politano L. Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. Hum Genet 2016;135:685-98.
15. Viggiano E, Picillo E, Ergoli M, Cirillo A, Del Gaudio S, Politano L. Skewed X-chromosome inactivation plays a crucial role in the onset of symptoms in carriers of Becker muscular dystrophy. J Gene Med 2017; doi: 10.1002/jgm.2952.
16. Winchester B, Young E, Geddes S, Genet S, Hurst J, Middleton-Price H, Williams N, Webb M, Habel A, Malcolm S. Female twin with Hunter disease due to nonrandom inactivation of the X-chromosome: a consequence of twinning. Am J Med Genet 1992;44:834-8.
17. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007;315:1143-7.
18. Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrere F, Blanche S, Audit M, Payen E, Leboulch P, l'Homme B, Bougneres P, Von Kalle C, Fischer A, Cavazzana-Calvo M, Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009;326:818-23.
19. Polito VA, Cosma MP. IDS crossing of the blood-brain barrier corrects CNS defects in MPSII mice. Am J Hum Genet 2009;85:296-301.
20. Cardone M, Polito VA, Pepe S, Mann L, D'Azzo A, Auricchio A, Ballabio A, Cosma MP. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum Mol Genet 2006;15:1225-36.
21. Maier EM, Kammerer S, Muntau AC, Wichers M, Braun A, Roscher AA. Symptoms in carriers of adrenoleukodystrophy relate to skewed X inactivation. Ann Neurol 2002;52:683-8.
22. Cerase A, Pintacuda G, Tattermusch A, Avner P. Xist localization and function: new insights from multiple levels. Genome Biol 2015;16:166.
23. Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY. Systematic discovery of Xist RNA binding proteins. Cell 2015;161:404-16.
24. McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, Sweredoski MJ, Shishkin AA, Su J, Lander ES, Hess S, Plath K, Guttman M. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015;521:232-6.
25. Minajigi A, Froberg JE, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, Lee JT. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015;349:aab2276.
26. Moindrot B, Cerase A, Coker H, Masui O, Grijzenhout A, Pintacuda G, Schermelleh L, Nesterova TB, Brockdorff N. A pooled shRNA screen identifies Rbm15, Spen, and Wtap as factors required for Xist RNA-mediated silencing. Cell Rep 2015;12:562-72.
27. Monfort A, Di Minin G, Postlmayr A, Freimann R, Arieti F, Thore S, Wutz A. Identification of Spen as a crucial factor for Xist function through forward genetic screening in haploid embryonic stem cells. Cell Rep 2015;12:554-61.
28. Almeida M, Pintacuda G, Masui O, Koseki Y, Gdula M, Cerase A, Brown D, Mould A, Innocent C, Nakayama M, Schermelleh L, Nesterova TB, Koseki H, Brockdorff N. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 2017;356:1081-4.
29. Sarma K, Cifuentes-Rojas C, Ergun A, Del Rosario A, Jeon Y, White F, Sadreyev R, Lee JT. ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell 2014;159:869-83.
30. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008;322:750-6.
31. Chaumeil J, Le Baccon P, Wutz A, Heard E. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 2006;20:2223-37.
32. Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science 2003;300:131-5.
33. Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL. Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 2001;107:727-38.
34. Fang J, Chen T, Chadwick B, Li E, Zhang Y. Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J Biol Chem 2004;279:52812-5.
35. de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, Koseki H, Brockdorff N. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell 2004;7:663-76.
36. Costanzi C, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 1998;393:599-601.
37. Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science 2007;315:1141-3.
38. Changolkar LN, Costanzi C, Leu NA, Chen D, McLaughlin KJ, Pehrson JR. Developmental changes in histone macroH2A1-mediated gene regulation. Mol Cell Biol 2007;27:2758-64.
39. Leeb M, Wutz A. Ring1B is crucial for the regulation of developmental control genes and PRC1 proteins but not X inactivation in embryonic cells. J Cell Biol 2007;178:219-29.
40. Schoeftner S, Sengupta AK, Kubicek S, Mechtler K, Spahn L, Koseki H, Jenuwein T, Wutz A. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J 2006;25:3110-22.
41. Zhang LF, Huynh KD, Lee JT. Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell 2007;129:693-706.
42. Csankovszki G, Nagy A, Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol 2001;153:773-84.
43. Sripathy S, Leko V, Adrianse RL, Loe T, Foss EJ, Dalrymple E, Lao U, Gatbonton-Schwager T, Carter KT, Payer B, Paddison PJ, Grady WM, Lee JT, Bartolomei MS, Bedalov A. Screen for reactivation of MeCP2 on the inactive X chromosome identifies the BMP/TGF-beta superfamily as a regulator of XIST expression. Proc Natl Acad Sci U S A 2017;114:1619-24.
44. Bhatnagar S, Zhu X, Ou J, Lin L, Chamberlain L, Zhu LJ, Wajapeyee N, Green MR. Genetic and pharmacological reactivation of the mammalian inactive X chromosome. Proc Natl Acad Sci U S A 2014;111:12591-8.
45. Navarro P, Chambers I, Karwacki-Neisius V, Chureau C, Morey C, Rougeulle C, Avner P. Molecular coupling of Xist regulation and pluripotency. Science 2008;321:1693-5.
46. Navarro P, Oldfield A, Legoupi J, Festuccia N, Dubois A, Attia M, Schoorlemmer J, Rougeulle C, Chambers I, Avner P. Molecular coupling of Tsix regulation and pluripotency. Nature 2010;468:457-60.
47. Donohoe ME, Silva SS, Pinter SF, Xu N, Lee JT. The pluripotency factor Oct4 interacts with Ctcf and also controls X-chromosome pairing and counting. Nature 2009;460:128-32.
48. Gontan C, Achame EM, Demmers J, Barakat TS, Rentmeester E, van IW, Grootegoed JA, Gribnau J. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature 2012;485:386-90.
49. Eggan K, Akutsu H, Hochedlinger K, Rideout W 3rd, Yanagimachi R, Jaenisch R. X-Chromosome inactivation in cloned mouse embryos. Science 2000;290:1578-81.
50. Do JT, Han DW, Gentile L, Sobek-Klocke I, Stehling M, Scholer HR. Enhanced reprogramming of Xist by induced upregulation of Tsix and Dnmt3a. Stem Cells 2008;26:2821-31.
51. Pasque V, Gillich A, Garrett N, Gurdon JB. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J 2011;30:2373-87.
52. Pasque V, Tchieu J, Karnik R, Uyeda M, Sadhu Dimashkie A, Case D, Papp B, Bonora G, Patel S, Ho R, Schmidt R, McKee R, Sado T, Tada T, Meissner A, Plath K. X chromosome reactivation dynamics reveal stages of reprogramming to pluripotency. Cell 2014;159:1681-97.
53. Shen Y, Matsuno Y, Fouse SD, Rao N, Root S, Xu R, Pellegrini M, Riggs AD, Fan G. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc Natl Acad Sci U S A 2008;105:4709-14.
54. Silva SS, Rowntree RK, Mekhoubad S, Lee JT. X-chromosome inactivation and epigenetic fluidity in human embryonic stem cells. Proc Natl Acad Sci U S A 2008;105:4820-5.
55. Bruck T, Benvenisty N. Meta-analysis of the heterogeneity of X chromosome inactivation in human pluripotent stem cells. Stem Cell Res 2011;6:187-93.
56. Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, Garitaonandia I, Muller FJ, Wang YC, Boscolo FS, Fakunle E, Dumevska B, Lee S, Park HS, Olee T, D'Lima DD, Semechkin R, Parast MM, Galat V, Laslett AL, Schmidt U, Keirstead HS, Loring JF, Laurent LC. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 2012;10:620-34.
57. Patel S, Bonora G, Sahakyan A, Kim R, Chronis C, Langerman J, Fitz-Gibbon S, Rubbi L, Skelton RJ, Ardehali R, Pellegrini M, Lowry WE, Clark AT, Plath K. Human embryonic stem cells do not change their X inactivation status during differentiation. Cell Rep 2017;18:54-67.
58. Anguera MC, Sadreyev R, Zhang Z, Szanto A, Payer B, Sheridan SD, Kwok S, Haggarty SJ, Sur M, Alvarez J, Gimelbrant A, Mitalipova M, Kirby JE, Lee JT. Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell 2012;11:75-90.
59. Mekhoubad S, Bock C, de Boer AS, Kiskinis E, Meissner A, Eggan K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 2012;10:595-609.
60. Okamoto I, Patrat C, Thepot D, Peynot N, Fauque P, Daniel N, Diabangouaya P, Wolf JP, Renard JP, Duranthon V, Heard E. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 2011;472:370-4.
61. Petropoulos S, Edsgard D, Reinius B, Deng Q, Panula SP, Codeluppi S, Reyes AP, Linnarsson S, Sandberg R, Lanner F. Single-cell RNA-Seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell 2016;167:285.
62. Sahakyan A, Kim R, Chronis C, Sabri S, Bonora G, Theunissen TW, Kuoy E, Langerman J, Clark AT, Jaenisch R, Plath K. Human naive pluripotent stem cells model X chromosome dampening and X inactivation. Cell Stem Cell 2017;20:87-101.
63. Mak W, Nesterova TB, de Napoles M, Appanah R, Yamanaka S, Otte AP, Brockdorff N. Reactivation of the paternal X chromosome in early mouse embryos. Science 2004;303:666-9.
64. Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 2004;303:644-9.
65. Chuva de Sousa Lopes SM, Hayashi K, Shovlin TC, Mifsud W, Surani MA, McLaren A. X chromosome activity in mouse XX primordial germ cells. PLoS Genet 2008;4:e30.
66. Sugimoto M, Abe K. X chromosome reactivation initiates in nascent primordial germ cells in mice. PLoS Genet 2007;3:e116.
67. de Napoles M, Nesterova T, Brockdorff N. Early loss of Xist RNA expression and inactive X chromosome associated chromatin modification in developing primordial germ cells. PLoS One 2007;2:e860.
68. Takagi N, Yoshida MA, Sugawara O, Sasaki M. Reversal of X-inactivation in female mouse somatic cells hybridized with murine teratocarcinoma stem cells in vitro. Cell 1983;34:1053-62.
69. Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr Biol 2001;11:1553-8.
70. Choi HW, Kim JS, Jang HJ, Choi S, Kim JH, Scholer HR, Do JT. Reestablishment of the inactive X chromosome to the ground state through cell fusion-induced reprogramming. Cell Mol Life Sci 2012;69:4067-77.
71. Silva J, Chambers I, Pollard S, Smith A. Nanog promotes transfer of pluripotency after cell fusion. Nature 2006;441:997-1001.
72. Okuyama K, Takagi N, Sasaki M. Sequential X-chromosome reactivation and inactivation in cell hybrids between murine embryonal carcinoma cells and female rat thymocytes. Exp Cell Res 1986;164:323-34.
73. Piccolo FM, Bagci H, Brown KE, Landeira D, Soza-Ried J, Feytout A, Mooijman D, Hajkova P, Leitch HG, Tada T, Kriaucionis S, Dawlaty MM, Jaenisch R, Merkenschlager M, Fisher AG. Different roles for Tet1 and Tet2 proteins in reprogramming-mediated erasure of imprints induced by EGC fusion. Mol Cell 2013;49:1023-33.
74. Tada M, Tada T, Lefebvre L, Barton SC, Surani MA. Embryonic germ cells induce epigenetic reprogramming of somatic nucleus in hybrid cells. EMBO J 1997;16:6510-20.
75. Pereira CF, Terranova R, Ryan NK, Santos J, Morris KJ, Cui W, Merkenschlager M, Fisher AG. Heterokaryon-based reprogramming of human B lymphocytes for pluripotency requires Oct4 but not Sox2. PLoS Genet 2008;4:e1000170.
76. Lee JT. Disruption of imprinted X inactivation by parent-of-origin effects at Tsix. Cell 2000;103:17-27.
77. Lee JT, Lu N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell 1999;99:47-57.
78. Sado T, Wang Z, Sasaki H, Li E. Regulation of imprinted X-chromosome inactivation in mice by Tsix. Development 2001;128:1275-86.
79. Yoshida I, Nishita Y, Mohandas TK, Takagi N. Reactivation of an inactive human X chromosome introduced into mouse embryonal carcinoma cells by microcell fusion with persistent expression of XIST. Exp Cell Res 1997;230:208-19.
81. Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature 1997;385:810-3.
82. Egli D, Sandler VM, Shinohara ML, Cantor H, Eggan K. Reprogramming after chromosome transfer into mouse blastomeres. Curr Biol 2009;19:1403-9.
83. Egli D, Rosains J, Birkhoff G, Eggan K. Developmental reprogramming after chromosome transfer into mitotic mouse zygotes. Nature 2007;447:679-85.
84. Egli D, Eggan K. Recipient cell nuclear factors are required for reprogramming by nuclear transfer. Development 2010;137:1953-63.
85. Kang E, Wu G, Ma H, Li Y, Tippner-Hedges R, Tachibana M, Sparman M, Wolf DP, Scholer HR, Mitalipov S. Nuclear reprogramming by interphase cytoplasm of two-cell mouse embryos. Nature 2014;509:101-4.
86. Egli D, Birkhoff G, Eggan K. Mediators of reprogramming: transcription factors and transitions through mitosis. Nat Rev Mol Cell Biol 2008;9:505-16.
87. Rideout WM 3rd, Eggan K, Jaenisch R. Nuclear cloning and epigenetic reprogramming of the genome. Science 2001;293:1093-8.
88. Inoue K, Kohda T, Lee J, Ogonuki N, Mochida K, Noguchi Y, Tanemura K, Kaneko-Ishino T, Ishino F, Ogura A. Faithful expression of imprinted genes in cloned mice. Science 2002;295:297.
89. Cantone I, Fisher AG. Epigenetic programming and reprogramming during development. Nat Struct Mol Biol 2013;20:282-9.
90. Nolen LD, Gao S, Han Z, Mann MR, Gie Chung Y, Otte AP, Bartolomei MS, Latham KE. X chromosome reactivation and regulation in cloned embryos. Dev Biol 2005;279:525-40.
91. Bao S, Miyoshi N, Okamoto I, Jenuwein T, Heard E, Azim Surani M. Initiation of epigenetic reprogramming of the X chromosome in somatic nuclei transplanted to a mouse oocyte. EMBO Rep 2005;6:748-54.
92. Inoue K, Kohda T, Sugimoto M, Sado T, Ogonuki N, Matoba S, Shiura H, Ikeda R, Mochida K, Fujii T, Sawai K, Otte AP, Tian XC, Yang X, Ishino F, Abe K, Ogura A. Impeding Xist expression from the active X chromosome improves mouse somatic cell nuclear transfer. Science 2010;330:496-9.
93. Matoba S, Inoue K, Kohda T, Sugimoto M, Mizutani E, Ogonuki N, Nakamura T, Abe K, Nakano T, Ishino F, Ogura A. RNAi-mediated knockdown of Xist can rescue the impaired postimplantation development of cloned mouse embryos. Proc Natl Acad Sci U S A 2011;108:20621-6.
94. Chadwick BP, Willard HF. Cell cycle-dependent localization of macroH2A in chromatin of the inactive X chromosome. J Cell Biol 2002;157:1113-23.
95. Hall LL, Byron M, Pageau G, Lawrence JB. AURKB-mediated effects on chromatin regulate binding versus release of XIST RNA to the inactive chromosome. J Cell Biol 2009;186:491-507.
96. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663-76.
97. Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, Plath K, Hochedlinger K. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007;1:55-70.
98. Payer B, Rosenberg M, Yamaji M, Yabuta Y, Koyanagi-Aoi M, Hayashi K, Yamanaka S, Saitou M, Lee JT. Tsix RNA and the germline factor, PRDM14, link X reactivation and stem cell reprogramming. Mol Cell 2013;52:805-18.
99. Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012;150:1209-22.
100. Yamaji M, Seki Y, Kurimoto K, Yabuta Y, Yuasa M, Shigeta M, Yamanaka K, Ohinata Y, Saitou M. Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat Genet 2008;40:1016-22.
101. Gillich A, Bao S, Surani MA. Reversion of mouse postimplantation epiblast stem cells to a naive pluripotent state by modulation of signalling pathways. Methods Mol Biol 2013;1074:15-29.
102. Ohhata T, Senner CE, Hemberger M, Wutz A. Lineage-specific function of the noncoding Tsix RNA for Xist repression and Xi reactivation in mice. Genes Dev 2011;25:1702-15.
103. Vallot C, Patrat C, Collier AJ, Huret C, Casanova M, Liyakat Ali TM, Tosolini M, Frydman N, Heard E, Rugg-Gunn PJ, Rougeulle C. XACT noncoding RNA competes with XIST in the control of X chromosome activity during human early development. Cell Stem Cell 2017;20:102-11.
104. Navarro P, Avner P. When X-inactivation meets pluripotency: an intimate rendezvous. FEBS Lett 2009;583:1721-7.
105. Takagi N, Abe K. Detrimental effects of two active X chromosomes on early mouse development. Development 1990;109:189-201.
106. Marahrens Y, Panning B, Dausman J, Strauss W, Jaenisch R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev 1997;11:156-66.
107. Vallot C, Ouimette JF, Makhlouf M, Feraud O, Pontis J, Come J, Martinat C, Bennaceur-Griscelli A, Lalande M, Rougeulle C. Erosion of X chromosome inactivation in human pluripotent cells initiates with XACT coating and depends on a specific heterochromatin landscape. Cell Stem Cell 2015; doi: 10.1016/j.stem.2015.03.016.
108. Dvash T, Lavon N, Fan G. Variations of X chromosome inactivation occur in early passages of female human embryonic stem cells. PLoS One 2010;5:e11330.
109. Vallot C, Huret C, Lesecque Y, Resch A, Oudrhiri N, Bennaceur-Griscelli A, Duret L, Rougeulle C. XACT, a long noncoding transcript coating the active X chromosome in human pluripotent cells. Nat Genet 2013;45:239-41.
110. Diaz Perez SV, Kim R, Li Z, Marquez VE, Patel S, Plath K, Clark AT. Derivation of new human embryonic stem cell lines reveals rapid epigenetic progression in vitro that can be prevented by chemical modification of chromatin. Hum Mol Genet 2012;21:751-64.
111. Wutz A. Epigenetic alterations in human pluripotent stem cells: a tale of two cultures. Cell Stem Cell 2012;11:9-15.
112. Lengner CJ, Gimelbrant AA, Erwin JA, Cheng AW, Guenther MG, Welstead GG, Alagappan R, Frampton GM, Xu P, Muffat J, Santagata S, Powers D, Barrett CB, Young RA, Lee JT, Jaenisch R, Mitalipova M. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell 2010;141:872-83.
113. Xie P, Ouyang Q, Leng L, Hu L, Cheng D, Tan Y, Lu G, Lin G. The dynamic changes of X chromosome inactivation during early culture of human embryonic stem cells. Stem Cell Res 2016;17:84-92.
114. Gafni O, Weinberger L, Mansour AA, Manor YS, Chomsky E, Ben-Yosef D, Kalma Y, Viukov S, Maza I, Zviran A, Rais Y, Shipony Z, Mukamel Z, Krupalnik V, Zerbib M, Geula S, Caspi I, Schneir D, Shwartz T, Gilad S, Amann-Zalcenstein D, Benjamin S, Amit I, Tanay A, Massarwa R, Novershtern N, Hanna JH. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013;504:282-6.
115. Ware CB, Nelson AM, Mecham B, Hesson J, Zhou W, Jonlin EC, Jimenez-Caliani AJ, Deng X, Cavanaugh C, Cook S, Tesar PJ, Okada J, Margaretha L, Sperber H, Choi M, Blau CA, Treuting PM, Hawkins RD, Cirulli V, Ruohola-Baker H. Derivation of naive human embryonic stem cells. Proc Natl Acad Sci U S A 2014;111:4484-9.
116. Theunissen TW, Powell BE, Wang H, Mitalipova M, Faddah DA, Reddy J, Fan ZP, Maetzel D, Ganz K, Shi L, Lungjangwa T, Imsoonthornruksa S, Stelzer Y, Rangarajan S, D'Alessio A, Zhang J, Gao Q, Dawlaty MM, Young RA, Gray NS, Jaenisch R. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell 2014;15:471-87.
117. Tchieu J, Kuoy E, Chin MH, Trinh H, Patterson M, Sherman SP, Aimiuwu O, Lindgren A, Hakimian S, Zack JA, Clark AT, Pyle AD, Lowry WE, Plath K. Female human iPSCs retain an inactive X chromosome. Cell Stem Cell 2010;7:329-42.
118. Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010;143:527-39.
119. Pomp O, Dreesen O, Leong DF, Meller-Pomp O, Tan TT, Zhou F, Colman A. Unexpected X chromosome skewing during culture and reprogramming of human somatic cells can be alleviated by exogenous telomerase. Cell Stem Cell 2011;9:156-65.
120. Tomoda K, Takahashi K, Leung K, Okada A, Narita M, Yamada NA, Eilertson KE, Tsang P, Baba S, White MP, Sami S, Srivastava D, Conklin BR, Panning B, Yamanaka S. Derivation conditions impact X-inactivation status in female human induced pluripotent stem cells. Cell Stem Cell 2012;11:91-9.
121. Kim KY, Hysolli E, Tanaka Y, Wang B, Jung YW, Pan X, Weissman SM, Park IH. X chromosome of female cells shows dynamic changes in status during human somatic cell reprogramming. Stem Cell Reports 2014;2:896-909.
122. Takashima Y, Guo G, Loos R, Nichols J, Ficz G, Krueger F, Oxley D, Santos F, Clarke J, Mansfield W, Reik W, Bertone P, Smith A. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 2014;158:1254-69.
123. Huang K, Maruyama T, Fan G. The naive state of human pluripotent stem cells: a synthesis of stem cell and preimplantation embryo transcriptome analyses. Cell Stem Cell 2014;15:410-5.
124. Brady JJ, Li M, Suthram S, Jiang H, Wong WH, Blau HM. Early role for IL-6 signalling during generation of induced pluripotent stem cells revealed by heterokaryon RNA-Seq. Nat Cell Biol 2013;15:1244-52.
125. Harris H, Watkins JF, Ford CE, Schoefl GI. Artificial heterokaryons of animal cells from different species. J Cell Sci 1966;1:1-30.
126. Cantone I, Bagci H, Dormann D, Dharmalingam G, Nesterova T, Brockdorff N, Rougeulle C, Vallot C, Heard E, Chaligne R, Merkenschlager M, Fisher AG. Ordered chromatin changes and human X chromosome reactivation by cell fusion-mediated pluripotent reprogramming. Nat Commun 2016;7:12354.
127. Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, Plath K, Guttman M. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013;341:1237973.
128. Smeets D, Markaki Y, Schmid VJ, Kraus F, Tattermusch A, Cerase A, Sterr M, Fiedler S, Demmerle J, Popken J, Leonhardt H, Brockdorff N, Cremer T, Schermelleh L, Cremer M. Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenetics Chromatin 2014;7:8.
129. Cantone I, Dharmalingam G, Chan YW, Kohler AC, Lenhard B, Merkenschlager M, Fisher AG. Allele-specific analysis of cell fusion-mediated pluripotent reprograming reveals distinct and predictive susceptibilities of human X-linked genes to reactivation. Genome Biol 2017;18:2.
130. DeBoever C, Li H, Jakubosky D, Benaglio P, Reyna J, Olson KM, Huang H, Biggs W, Sandoval E, D'Antonio M, Jepsen K, Matsui H, Arias A, Ren B, Nariai N, Smith EN, D'Antonio-Chronowska A, Farley EK, Frazer KA. Large-scale profiling reveals the influence of genetic variation on gene expression in human induced pluripotent stem cells. Cell Stem Cell 2017;20:533-46.e7.
131. Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, Dekker J. Structural organization of the inactive X chromosome in the mouse. Nature 2016; doi: 10.1038/nature18589.
132. Berletch JB, Yang F, Xu J, Carrel L, Disteche CM. Genes that escape from X inactivation. Hum Genet 2011;130:237-45.
133. Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D, Starmer J, Mieczkowski P, Crawford GE, Magnuson T. Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 2012;151:951-63.
134. Berletch JB, Ma W, Yang F, Shendure J, Noble WS, Disteche CM, Deng X. Escape from X inactivation varies in mouse tissues. PLoS Genet 2015;11:e1005079.
135. Chaligne R, Popova T, Mendoza-Parra MA, Saleem MA, Gentien D, Ban K, Piolot T, Leroy O, Mariani O, Gronemeyer H, Vincent-Salomon A, Stern MH, Heard E. The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res 2015;25:488-503.
136. Kang J, Lee HJ, Kim J, Lee JJ, Maeng LS. Dysregulation of X chromosome inactivation in high grade ovarian serous adenocarcinoma. PLoS One 2015;10:e0118927.
137. Pageau GJ, Hall LL, Ganesan S, Livingston DM, Lawrence JB. The disappearing Barr body in breast and ovarian cancers. Nat Rev Cancer 2007;7:628-33.
138. Bennett-Baker PE, Wilkowski J, Burke DT. Age-associated activation of epigenetically repressed genes in the mouse. Genetics 2003;165:2055-62.
139. Ostan R, Monti D, Gueresi P, Bussolotto M, Franceschi C, Baggio G. Gender, aging and longevity in humans: an update of an intriguing/neglected scenario paving the way to a gender-specific medicine. Clin Sci (Lond) 2016;130:1711-25.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Cantone I. Reversal of X chromosome inactivation: lessons from pluripotent reprogramming of mouse and human somatic cells. J Transl Genet Genom 2017;1:1-14. http://dx.doi.org/10.20517/jtgg.2017.19
AMA Style
Cantone I. Reversal of X chromosome inactivation: lessons from pluripotent reprogramming of mouse and human somatic cells. Journal of Translational Genetics and Genomics. 2017; 1: 1-14. http://dx.doi.org/10.20517/jtgg.2017.19
Chicago/Turabian Style
Cantone, Irene. 2017. "Reversal of X chromosome inactivation: lessons from pluripotent reprogramming of mouse and human somatic cells" Journal of Translational Genetics and Genomics. 1: 1-14. http://dx.doi.org/10.20517/jtgg.2017.19
ACS Style
Cantone, I. Reversal of X chromosome inactivation: lessons from pluripotent reprogramming of mouse and human somatic cells. J. Transl. Genet. Genom. 2017, 1, 1-14. http://dx.doi.org/10.20517/jtgg.2017.19
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.