
Gene dosage defects in primary immunodeficiencies and related disorders: a pilot study

To the editor
Detection of structural variations or copy number variants (CNVs) is a critical component of diagnosing many genetic disorders especially when the causative mutation(s) cannot be identified in dominant, recessive and X-linked disorders by Sanger or next generation sequencing. Fluorescence in situ hybridization, whole genome copy number microarrays, next generation sequencing, Sanger sequencing, multiplex ligation-dependent probe amplification, and quantitative real-time polymerase chain reaction (PCR) may miss potential pathogenic structural variations that range in size larger than 500 bp or includes at least one exon. Moreover, CNV calls extracted from whole exome sequencing remain a challenge for clinical genetic testing laboratories due to high false positive rates. To overcome these technical challenges, we designed a custom high-resolution targeted exon-centric microarray (ECM) that has 1508 disease-causing genes including complementary genes to our laboratory’s immunodeficiency focused next-generation sequencing (NGS) panels. The combination of targeting rare immunodeficiency genes with large case load of clinical NGS testing for rare immunological and related disorders allowed for the opportunity to discover novel CNVs and expand our understanding of disease mechanisms in this group of genetically heterogeneous disorders. We have been routinely performing the ECM as a clinical assay and herein report a number of novel CNVs detected in this group of disorders for which intragenic deletions are not a commonly reported etiology. We followed the diagnostic algorithm illustrated in [Figure 1], to increase the diagnostic yield in patients in this group of disorders, but albeit is not limited to these disorders and is recommended for Mendelian disorders because the mutation spectrum of many genes remain understudied and deletions and duplications may represent a significant percentage of pathogenic changes.
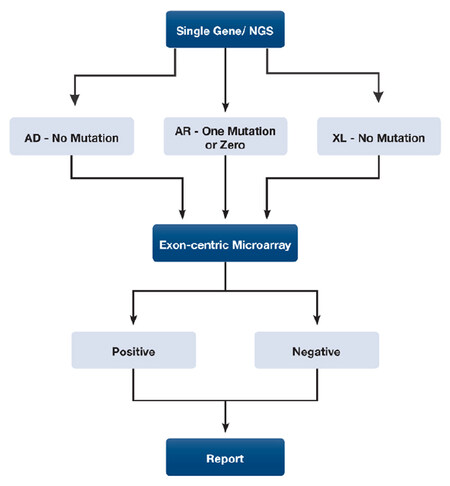
Figure 1. CNV testing algorithm for Mendelian disorders. The diagram demonstrates the diagnostic work-up of a Mendelian disorder using next generation sequencing as the first step followed by deletion/duplication testing to find an allele or second allele. CNV: copy number variant; NGS: next-generation sequencing
Clinical array comparative genomic hybridization was performed on 1048 patient samples referred to the Molecular Genetics Laboratory at Cincinnati Children’s Hospital Medical Center (CCHMC). ECM analysis was performed on only the gene(s) of interest. Reference DNA was obtained from Promega (Madison, WI). This study was approved by the Institutional Review Board at CCHMC.
The custom 4X180K microarray was developed on the Agilent Technologies (Santa Clara, CA) platform using the Genefficiency service (Oxford Gene Technology, Oxford, UK). OGT used proprietary ink-jet in situ printer technology (IJISS) developed by Rosetta InPharmatics (Kirkland, WA) and Agilent Technologies that allows in situ synthesis of long oligonucleotides. The probes were 60 bp in length and annotated against NCBI build 37 (UCSC hg19, March 2006). Included were 364,591 probes that targeted 1508 genes (exons and introns), up to 300 bp flanking the exon/intron boundaries and backbone coverage.
DNA was extracted from whole blood collected in EDTA (purple-top) collection tubes using the Puregene DNA extraction kit (Qiagen, Valencia, CA) or the Chemagic magnetic separation module I (PerkinElmer, Baesweiler, Germany) according to the manufacturer’s recommendations. Comparative genomic hybridization was performed following the manufacturer’s protocol (Agilent Technologies, Santa Clara, CA). The reference DNA was used from two pools (male and female) from normal individuals, run as a same-sex control. DNA was sonicated using a Branson Sonifier 450 with cup horn (Danbury, CT) and visualized on a two-percent agarose gel prior to labeling, as a quality control measure. Each patient and reference DNA was labeled with Cy3 and Cy5 9-mer primers, respectively. Purification of labeled products, hybridization, and post-wash of the array was carried out according to Agilent’s recommendation and with their proprietary solutions. Microarray slides were scanned with Innopsys Innoscan 710 and raw data was generated with extraction software (Chicago, Illinois).
CytoSure Interpret software version 4.4 (OGT) was used for analysis of microarray data (referred to as CytoSure). The program uses the circular binary segmentation algorithm to generate segments along the chromosomes that have similar copy number relative to reference chromosome. Averaging of the segments is with median value of all segments on a chromosome as the baseline. Deletion or duplication CNV calls were made using the log2 ratio of each segment with a minimum of 4 unique and consequent probes. The threshold factor for deletions was set as a log2 ratio of -0.6 that is less stringent than the theoretical log2 score of -1 for heterozygous deletions [heterozygous deletion log2(1/2) = -1; no change in allele number log2(2/2) = 0; heterozygous duplication log2(3/2) = 0.59; hemizygous deletion log2(0/1)= -1; homozygous deletion log2(0/2)= -2]. The software uses the standard deviation of the log2 ratio to calculate a deviation log ratio (DLR), which is used as a quality control check. A DLR of 0.08-0.19 is acceptable, 0.20-0.29 is borderline, and ≥ 0.30 is rejected. The DLR for all microarrays shown was scored by this scale. Data was analyzed only for the gene(s) ordered for testing.
We used the high-resolution ECM to process 1048 of samples that were reflex testing from negative or equivocal targeted NGS panel testing or ordered for single gene assessment. We identified 23 CNVs in rare immunodeficiency and related genes [Table 1]. Of the 23 CNVs detected, 21 deletions were identified. Significantly, of these 23 CNVs, 11 (48%) of them were novel, for which intragenic deletions and duplications are not a commonly reported etiology, either due to the low mutation burden of these events or the lack of recognition by an appropriate testing methodology. Six of the 23 CNVs in immunodeficiency and related genes were identified in dominantly inherited genes (26%), 7 hemizygous CNVs were identified in male patients in X-linked recessive genes (30%), 5 CNVs were identified as the second allele in 3 autosomal recessive genes (17%), and surprisingly, 3 homozygous deletions were identified (13%).
List of patients with CNVs identified by exon-centric microarray testing
| Patient | Gene | OMIM phenotype: inheritance | CNV coordinates (min) | Min size (Max size) | Min region affected | Protein domain affected | Sequencing results | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | FAS | Autoimmune lymphoproliferative syndrome: AD | arr[hg19] 10q23.31 (90,770,398-90,777,298) x1 | 6.9 Kb (53.59 Kb) | exons 6-9 | Death-like | No variant identified | This study |
| 2 | FAS | Autoimmune lymphoproliferative syndrome: AD | arr[hg19] 10q23.31 (90,748,451-91,780,017) x1 | 1.03 Mb (1.19 Mb) | whole gene | No variant identified | This study | |
| 3^ | FAS | Autoimmune lymphoproliferative syndrome: AD | arr[hg19] 10q23.21 (90774157-90774381) x0 | 224bp (403bp) | exon 9 | Death-like | No variant identified | This study |
| 4 | RPS26 | Diamond-Blackfan anemia: AD | arr[hg19] 12q13.2q13.3 (56,175,892-56,691,772) x1 | 515.88 Kb (688.55 Kb) | whole gene | N/A | [6] | |
| 5 | RPL35A | Diamond-Blackfan anemia: AD | arr[hg19] 3q29 (197,456,033-197,844,960) x1 | 388.93 Kb (855.23 Kb) | whole gene | N/A | [6] | |
| 6 | TERT | Dyskeratosis congenita: AD | arr[hg19] 5p15.33 (1,226,629-1,286,091) x1 | 59.46 Kb (60.16 Kb) | exon 3-16 | Reverse transcriptase | No variant identified | This study |
| 7* | WAS | Wiskot-Aldrich syndrome: XLR | arr[hg19] Xp11.23 (48,533,049-48,542,539) x0 | 9.49 Kb (147.2 Kb) | exon 1 | EVH1 | No variant identified | This study |
| 8* | WAS | Wiskot-Aldrich syndrome: XLR | arr[hg19] Xp11.23 (48,533,302-48,613,923) x0 | 80.62 Kb (110.45 Kb) | whole gene | No variant identified | [7] | |
| 9 | WAS | Wiskot-Aldrich syndrome: XLR | arr[hg19] Xp11.23 (48,533,049-48,550,909) x0 | 17.86 Kb (220.16 Kb) | whole gene | No variant identified | [7] | |
| 10 | WAS | Wiskot-Aldrich syndrome: XLR | arr[hg19] Xp11.23 (48,533,049-48,551,398) x1 | 18.35 Kb (282.83 Kb) | whole gene | No variant identified | [7] | |
| 11 | SH2D1A | X-linked recessive lymphoproliferative disorder: XLR | arr[hg19] Xq25 (123,481,887-123,501,318) x0 | 19.43 Kb (20.58 Kb) | exon 2 | SH2 | No variant identified | [8,9] |
| 12 | XIAP | X-linked recessive lymphoproliferative disorder: XLR | arr[hg19] Xq25 (123,007,906-123,021,550) x0 | 13.64 Kb (15.61 Kb) | exon 2 | Baculoviral inhibition of apoptosis protein repeat | No variant identified | This study |
| 13 | MAGT1 | Immunodeficiency: XLR | arr[hg19]Xq21.1 (77,088,396-77,150,089) x0 | 63 Kb (63 Kb) | Exons 2-7 | Oligosaccharyl transferase complex | No variant identified | This study |
| 14 | IL7R | Severe combined immunodeficiency, T-cell −, B-cell +: AR | arr[hg19] 5p13.2 (35,867,205-35,867,604) x1 | 399 bp (3.94 Kb) | exon 3 | c.669_688del20 (p.S223fs) | [10] | |
| 15 | FANCA | Fanconi Anemia: AR | arr[hg19] 16q24.3 (89,880,335-89,928,824) x0 | 48.49 Kb (107.96 Kb) | exons 1-3 | No variant identified | [11] | |
| 16 | FANCA | Fanconi Anemia: AR | arr[hg19] 16p24.3 (89,874,512-89,884,589) x1 | 10.08kb (56.89 Kb) | exons 1-6 | c.862G>T(p.E288X) | [12] | |
| 17 | FANCA | Fanconi Anemia: AR | arr[hg19] 16q24.3 (89,880,335-89,928,824) x1 | 48.49 Kb (107.96 Kb) | exons 1-3 | c.3918dupT(p.Q1307fs) | [11] | |
| 18 | FANCC | Fanconi Anemia: AR | arr[hg19] 9q22.32 (98,007,056-98,011,765) x1 | 4.71 Kb (5.77 Kb) | exons 2-3 | c.1642C>T(p.R548*) | [13,14] | |
| 19 | RAD51C | Fanconi Anemia: AR | arr[hg19] 17q22 (56,765,347-56,790,584) x3 | 25.24 Kb (156.62 Kb) | exons 1-5 | DNA recombination and repair, ATP binding domain | No variant identified | This study |
| 20 | LRBA | Immunodeficiency: AR | arr[hg19] 4q31.3 (151,198,604-151,199,333) x0 | 729 bp (7.59 Kb) | exon 57 | WD40 repeat | No variant identified | This study |
| 21^ | STXBP2 | Hemophagocytic lymphohistiocytosis: AR | arr[hg19] 19p13.2 (7,706,766-7,781,506) x0 | 74.74 Kb (426.44 Kb) | exons 8-19 | Sec-like protein | No variant identified | This study |
| 22 | RAB27A | Griscelli syndrome, type 2: AR | arr[hg19] 15q21.3 (55,499,521-55,562,873) x1 | 63.35 Kb (67.75 Kb) | exons 1-5 | GTPase | No variant identified | [15] |
| 23 | RAB27A | Griscelli syndrome, type 2: AR | arr[hg19] 15q21.3 (55,514,892-55,550,411) x3 | 35.52 Kb (40.86 Kb) | exons 2-5 | GTPase | No variant identified | This study |
Meaningfully, we identified 3 novel CNVs in the FAS gene. Heterozygous mutations in the FAS gene cause autoimmune lymphoproliferative syndrome (ALPS), an autosomal dominant disorder, where only three CNVs have been previously described[1-4]. In patient 1, we identified a 6.9 Kb heterozygous deletion that encompassed exons 6-9 and included the intracellular domain and the death domain of the FAS protein. Single gene CNV assessment of FAS was ordered for this patient after negative sequencing of FAS, FASLG, and CASP10 genes. This patient’s phenotype included splenomegaly, thrombocytopenia, history or acute cerebellitis and failure to thrive. Flow cytometry studies from an external laboratory identified an increased population of CD4/CD8 double negative T cells and reduced FAS protein expression. Similarly, in patient 2 and 3, we identified a 1.03 Mb whole gene heterozygous deletion and a 224 bp homozygous deletion in exon 9, respectively. ECM testing was ordered as reflex testing from Sanger sequencing in all 3 patients. The identification of the FAS gene deletions in these patients confirmed a diagnosis of ALPS. In addition, the CNV mutation spectrum of FAS was doubled, showing that ECM is a powerful tool for CNV discovery in rare Mendelian disorders.
Other rare hematology and immunodeficiency genes for which novel CNVs were identified included TERT, WAS, XIAP, MAGT1, RAD51C, LRBA, STXBP2, and RAB27A. Deletions in X-linked genes showed high clinical utility in males due to their hemizygous status. In several cases, a lack of amplification by PCR suggested a deletion, but results were inconclusive in clinical interpretation due to lack of direct evidence and insufficient controls. WAS-related disorders are X-linked recessive and include Wiskott-Aldrich syndrome, X-linked thrombocytopenia, and X-linked congenital neutropenia. Sequencing of the WAS gene identifies up to 95% of pathogenic variants and the remaining 5% of variants are detectable by deletion/duplication testing and 15 gross deletions have been reported. We identified 3 CNVs in 3 male patients involving the WAS gene. In patient 7, we identified a previously unreported 9.49 Kb intragenic deletion of exon 1, and in patients 8, 9 and 10, we identified previously reported whole gene deletions in WAS of size 80.62 Kb, 17.86 Kb, and 18.35 Kb, respectively. Autosomal dominant mutations in TERT cause dyskeratosis congenital, a rare multisystem disorder caused by defective telomere maintenance. We identified a novel 59.46 Kb heterozygous deletion of exons 3-16 and included the reverse transcriptase domain of the TERT protein, the catalytic component of the enzyme. To date, only one CNV in TERT had been identified in a family with adult-onset pulmonary fibrosis[4]. In addition, the LRBA protein is involved in regulating recycling of the cytotoxic T lymphocytes-associated protein 4, which helps regulate autoimmune responses. Biallelic deleterious mutations in LRBA have been associated with common variable immunodeficiency type 8 and to date, only 6 mutations have been identified in LRBA. The biological role of LRBA is currently being elucidated and recent publications have suggested targeted therapies for patients with LRBA deficiency[5]. We identified a novel 729 bp homozygous deletion of exon 57 of LRBA in a patient from consanguineous parents.
In our cohort, we recognized a high diagnostic yield for the ECM assays for immunodeficiencies and related conditions. However, functional studies will be necessary to confirm the causative relationship to disease. This report emphasizes the importance of appropriate ECM testing for immunodeficiency disorders and helps further define gene mutation spectra and our understanding of disease mechanisms in this understudied group of genetically heterogeneous disorders.
Declarations
Authors’ contributionsConceptualized and initiated the study, collected, analyzed, and interpreted the data, wrote and reviewed the manuscript: C.A. Valencia
Analyzed and interpreted the data, wrote and reviewed the manuscript: L. Dyer, X. Li, K. Zhang
Developed the test and reviewed the manuscript: A. Mathur, J. Denton
Performed the experiments, collected, analyzed and interpreted the data and reviewed the manuscript: B. Jones, E. Liston, D. Hilton
Approved the final manuscript as submitted and agree to be accountable for all aspects of the work: all authors
Financial support and sponsorshipThe study was supported by the clinical molecular genetics laboratory at CCHMC.
Conflicts of interestThe authors declare that they have no relevant conflicts of interest.
Patient consentNot applicable.
Ethics approvalThis study was approved by the Institutional Review Board at Cincinnati Children’s Hospital Medical Center.
Copyright© The Author(s) 2017.
REFERENCES
1. van der Werff ten Bosch JE, Demanet C, Balduck N, Bakkus MH, De Raeve H, Desprechins B, Otten J, Thielemans K. The use of the anti-malaria drug Fansidar (pyrimethamine and sulphadoxine) in the treatment of a patient with autoimmune lymphoproliferative syndrome and Fas deficiency. Br J Haematol 1998;102:578-81.
2. Magerus-Chatinet A, Neven B, Stolzenberg MC, Daussy C, Arkwright PD, Lanzarotti N, Schaffner C, Cluet-Dennetiere S, Haerynck F, Michel G, Bole-Feysot C, Zarhrate M, Radford-Weiss I, Romana SP, Picard C, Fischer A, Rieux-Laucat F. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest 2011;121:106-12.
3. Pensati L, Costanzo A, Ianni A, Accapezzato D, Iorio R, Natoli G, Nisini R, Almerighi C, Balsano C, Vajro P, Vegnente A, Levrero M. Fas/Apo1 mutations and autoimmune lymphoproliferative syndrome in a patient with type 2 autoimmune hepatitis. Gastroenterology 1997;113:1384-9.
4. Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A 2007;104:7552-7.
5. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, Zhang Y, Liu Z, Fritz JM, Marsh R, Husami A, Kissell D, Nortman S, Chaturvedi V, Haines H, Young LR, Mo J, Filipovich AH, Bleesing JJ, Mustillo P, Stephens M, Rueda CM, Chougnet CA, Hoebe K, McElwee J, Hughes JD, Karakoc-Aydiner E, Matthews HF, Price S, Su HC, Rao VK, Lenardo MJ, Jordan MB. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015;349:436-40.
6. Quarello P, Garelli E, Brusco A, Carando A, Mancini C, Pappi P, Vinti L, Svahn J, Dianzani I, Ramenghi U. High frequency of ribosomal protein gene deletions in Italian Diamond-Blackfan anemia patients detected by multiplex ligation-dependent probe amplification assay. Haematologica 2012;97:1813-7.
7. Imai K, Morio T, Zhu Y, Jin Y, Itoh S, Kajiwara M, Yata J-I, Mizutani S, Ochs HD, Nonoyama S. Clinical course of patients with WASP gene mutations. Blood 2004;103:456-64.
8. Yin L, Ferrand V, Lavoué MF, Hayoz D, Philippe N, Souillet G, Seri M, Giacchino R, Castagnola E, Hodgson S, Sylla BS, Romeo G. SH2D1A mutation analysis for diagnosis of XLP in typical and atypical patients. Hum Genet 1999;105:501-5.
9. Sumegi J, Huang D, Lanyi A, Davis JD, Seemayer TA, Maeda A, Klein G, Seri M, Wakiguchi H, Purtilo DT, Gross TG. Correlation of mutations of the SH2D1A gene and epstein-barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood 2000;96:3118-25.
10. Bayer DK, Martinez CA, Sorte HS, Forbes LR, Demmler-Harrison GJ, Hanson IC, Pearson NM, Noroski LM, Zaki SR, Bellini WJ, Leduc MS, Yang Y, Eng CM, Patel A, Rodningen OK, Muzny DM, Gibbs RA, Campbell IM, Shaw CA, Baker MW, Zhang V, Lupski JR, Orange JS, Seeborg FO, Stray-Pedersen A. Vaccine-associated varicella and rubella infections in severe combined immunodeficiency with isolated CD4 lymphocytopenia and mutations in IL7R detected by tandem whole exome sequencing and chromosomal microarray. Clin Exp Immunol 2014;178:459-69.
11. Castella M, Pujol R, Callén E, Trujillo JP, Casado JA, Gille H, Lach FP, Auerbach AD, Schindler D, Benítez J, Porto B, Ferro T, Mu-oz A, Sevilla J, Madero L, Cela E, Beléndez C, de Heredia CD, Olivé T, de Toledo JS, Badell I, Torrent M, Estella J, Dasí A, Rodríguez-Villa A, Gómez P, Barbot J, Tapia M, Molinés A, Figuera A, Bueren JA, Surrallés J. Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood 2011;117:3759-69.
12. Levran O, Diotti R, Pujara K, Batish SD, Hanenberg H, Auerbach AD. Spectrum of sequence variations in the FANCA gene: an International Fanconi Anemia Registry (IFAR) study. Hum Mutat 2005;25:142-9.
13. Lo ten Foe JR, Barel MT, Thuss P, Digweed M, Arwert F, Joenje H. Sequence variations in the Fanconi anaemia gene, FAC: pathogenicity of 1806insA and R548X and recognition of D195V as a polymorphic variant. Hum Genet 1996;98:522-3.
14. Hartmann L, Neveling K, Borkens S, Schneider H, Freund M, Grassman E, Theiss S, Wawer A, Burdach S, Auerbach AD, Schindler D, Hanenberg H, Schaal H. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am J Hum Genet 2010;87:480-93.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Dyer L, Li X, Denton J, Jones B, Liston E, Hilton D, Mathur A, Zhang K, Valencia CA. Gene dosage defects in primary immunodeficiencies and related disorders: a pilot study. J Transl Genet Genom 2017;1:23-7. http://dx.doi.org/10.20517/jtgg.2017.04
AMA Style
Dyer L, Li X, Denton J, Jones B, Liston E, Hilton D, Mathur A, Zhang K, Valencia CA. Gene dosage defects in primary immunodeficiencies and related disorders: a pilot study. Journal of Translational Genetics and Genomics. 2017; 1: 23-7. http://dx.doi.org/10.20517/jtgg.2017.04
Chicago/Turabian Style
Dyer, Lisa, Xia Li, James Denton, Brittany Jones, Emily Liston, Danielle Hilton, Abhinav Mathur, Kejian Zhang, C. Alexander Valencia. 2017. "Gene dosage defects in primary immunodeficiencies and related disorders: a pilot study" Journal of Translational Genetics and Genomics. 1: 23-7. http://dx.doi.org/10.20517/jtgg.2017.04
ACS Style
Dyer, L.; Li X.; Denton J.; Jones B.; Liston E.; Hilton D.; Mathur A.; Zhang K.; Valencia CA. Gene dosage defects in primary immunodeficiencies and related disorders: a pilot study. J. Transl. Genet. Genom. 2017, 1, 23-7. http://dx.doi.org/10.20517/jtgg.2017.04
About This Article
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.