
MERRF and MELAS: current gene therapy trends and approaches

Abstract
The mitochondrion is a unique organelle that predominantly functions to produce useful cellular energy in the form of adenosine triphosphate (ATP). Unlike other non-nuclear eukaryotic organelles (with the exception of chloroplasts), mitochondria have two lipid membranes that enclose their own mitochondrial DNA (mtDNA) and ribosomes for protein production. Similar to nuclear DNA, mtDNA is equally susceptible to mutations that may be classified as either pathogenic or nonpathogenic. Myoclonic Epilepsy with Ragged Red Fibers (MERRF) and Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes (MELAS) are mitochondrial diseases originating from pathogenic point mutations located within mtDNA. Currently, there is no cure and patient care primarily focuses on treating each disease’s associated symptoms. When considering the multiple barriers existing between the extracellular surface of the plasma membrane and the location of the mtDNA within the mitochondrial matrix, developing a pharmacological therapeutic that can both overcome these barriers and correct an mtDNA causing mitochondrial disease remains difficult at best. Interestingly, the field of gene therapy may provide an opportunity for effective therapeutic intervention by introducing a genetic payload (to a particular cellular gene) to induce the correction. This review primarily focuses on understanding the principles of mitochondrial biology leading to the mtDNA diseases, MERRF and MELAS, while providing a landscape perspective of gene therapy research devoted to curing these diseases.
Keywords
Introduction to mitochondrial biology
Mitochondrial structure and function
The scientist, Ivan Wallin, stated that mitochondria had originated as endosymbionts in 1927[1]. Since each component of a mitochondrion is unique onto itself, its ancestral roots are likely to have originated from an “alpha-proteobacteria (gender: Rickettsia) that would have been engulfed by a heterotrophic host cell, probably an archeobacterium, 1.5 billion years ago” as proposed by Lynn Margulis[2]. In Margulis’ book, Origin of Eukaryotic Cells, “symbiosis usually means two or more organisms with distinct evolutionary histories derive benefit from highly specific associations with each other”[3]. While this notion may have been initially controversial, it has since been postulated that symbiosis is a necessary component of evolution. Moreover, scientific evidence has emerged to support the endosymbiotic origin of mitochondria including: free-living form of early mitochondria, mitochondrial DNA and structure, as well as the relegation of mitochondrial metabolic processes to nuclear regulation[3].
The structure of the mitochondrion is quite unique from the remaining membrane-bounded eukaryotic organelles. Its composition consists of an outer mitochondrial membrane, an inter-membrane space, an inner mitochondrial membrane, and a mitochondrial matrix (containing the mtDNA and ribosomes). Due to the four hydrocarbon tailed phospholipid, cardiolipin, the inner mitochondrial membrane is able to form cristae structures that are resultant from membrane in-folding[4]. In 1988, Robin and Wong[5] had determined that mitochondrial statistical information (number of mitochondria/cell and number of mtDNA/cell) varied between the following mammalian cell lines: rabbit lung macrophages, rabbit peritoneal macrophages, mouse LA9 fibroblasts, human lung fibroblasts and rat L-8 skeletal muscle cells. As per their analysis, the following statistical measures were obtained: the amount of mitochondrial DNA per mitochondrion remained the same in all cell varieties, the amount of mitochondrial DNA molecules per cell experienced an eight-fold difference between the different cell types, while the virtual number of mitochondria per cell also varied[5]. Therefore, dependent on the cell type, the morphology of mitochondria can vary greatly, appearing as either distinct entities ranging from 1 to 4 µm in length, or as part of an organized network[6,7]. As an interesting point, the extent of mitochondrial morphology is dependent on the balance between mitochondrial fusion and fission[6]. Furthermore, these events have the potential to be influenced by the mitochondrial membrane mechanical characteristics[8].
Resultant of its unique structure, the mitochondrion serves as a crucial cellular metabolism component. Mitochondria are well-defined as the powerhouse of the eukaryotic cell, but in addition to its famous oxidative phosphorylation role, they participate in: the creation of iron-sulfur (Fe-S) clusters, β-oxidation of fatty acids, synthesis of heme prosthetic groups, steroidogenesis (dependent on the cell variety), the urea cycle, as well as the homeostatic maintenance of calcium[2]. Mitochondria contribute greatly to the generation of free radicals, and have a participatory role in inflammation and innate immunity[4]. Moreover, mitochondria serve as a component for critical cellular signals relating to either its survival or death[2]. A summary of mitochondrial functions, with their associated purpose, appears in Table 1[2,4,9-17].
Notable mitochondrial functions with its associated purpose (in addition to oxidative phosphorylation)
| Mitochondrial function | Purpose |
|---|---|
| Formation of Iron-Sulfur (Fe-S) clusters[2] | Small prosthetic groups that are inorganic and composed of iron-sulfur clusters[9] Role in electron transfer, binding and activation of substrates, redox catalysis, DNA replication, DNA repair, gene expression regulation, and modification of tRNAs[9] |
| β-oxidation of fatty acids[2] | Functions to drive oxidative phosphorylation and supply acetyl-CoA for ketogenesis[10] |
| Synthesis of heme prosthetic groups[2] | Production of heme through a complex biochemical process occurring in cells[11] |
| Steroidogenesis[2] | Production of steroid hormones from cholesterol[12] |
| Urea cycle[2] | Production of urea from ammonia[13] |
| Homeostatic maintenance of calcium[2] | Responsible for maintenance of calcium homeostasis required for biological processes[14] |
| Generation of free radicals[4] | Production of toxic byproducts resultant from mitochondrial energetic processes (i.e., oxidative phosphorylation)[15] |
| Inflammation and innate immunity[4] | Mitochondria serve as a hub for innate immune signaling[16] Mitochondrial damage leads to inflammation by releasing mitochondrial alarmins[16] |
| Cellular signals associated with cell survival and death[2] | Mitochondrial participation in the regulation of cell survival and cell death induced by oxidative stress[17] |
Mitochondrial DNA
While studying chick embryo mitochondria, mtDNA was discovered in 1963 by Nass and Nass[18]. Located inside of the matrix, mtDNA is a critical component of mitochondrial processes. Seemingly, the circular nature of the mitochondrial genome is reminiscent of its bacterial origins, and interestingly, its composition varies according to the nucleotide densities of its component strands. Therefore, each strand of the circular molecule is denoted as either the heavy (predominantly composed of guanine) or the light (predominantly composed of cytosine) strand[2].
Unlike nuclear DNA, mtDNA follows a non-mendelian transmission pattern and is inherited maternally via the oocyte (despite the postulation of a possible chance of paternal inheritance). In 1981, Anderson et al.[19] had initially determined the sequence of the human mitochondrial genome, as well as provided an explanation of the sequence in relation to the arrangement and expression of its genes, which was subsequently revised in 1999[20]. Human mtDNA is organized as a circular double-stranded molecule that is 16,569 base pairs in length and has a unique mitochondrial genetic code (that is different from the nuclear genetic code) due to certain codons that represent alternative amino acids or stop codons[2]. Similar to nuclear DNA, mitochondrial DNA is capable of undergoing the following processes: DNA replication, RNA transcription, protein translation, DNA repair, and DNA recombination.
The mitochondrial genome [Figure 1] is highly efficient, containing no introns and a small portion of noncoding DNA[2]. Interestingly, mtDNA’s unique noncoding region is defined as the displacement loop (D-loop), which is approximately 1118 base pairs in length and exists approximately from nucleotides 577 to 16,028[21]. This region contains transcription promoters and a replication origin defined respectively as: the heavy strand promoter (HSP), the light strand promoter (LSP) and the heavy strand replication origin (OH)[2,21]. An alternative replication element, the light strand replication origin (OL), is situated externally to this region[2,21]. In addition, the D-loop segment is triple-stranded and contains a unique 650 nucleotide strand located between both the heavy and light strands, named 7S DNA, as determined by its sedimentation attributes[2,22]. Within the D-loop, 7S DNA is bound to the light strand, which displaces the heavy strand[23]. While the entire function of 7S DNA is not completely defined, it has been hypothesized that it participates in mtDNA transcription and replication[2].
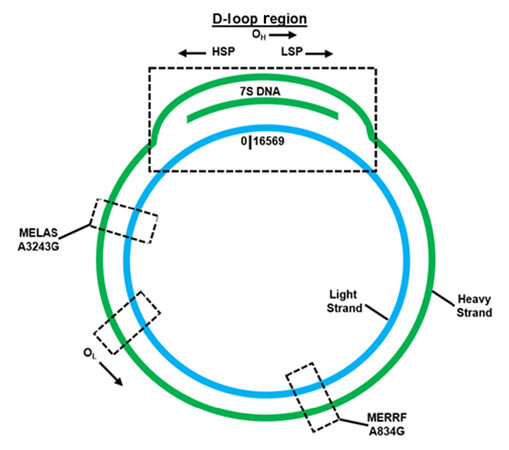
Figure 1. Overview of critical regulation, MERRF, and MELAS features located within the mitochondrial genome
DNA sequencing analysis has revealed unique regions on both strands of the human mitochondrial genome coding for 22 transfer RNAs (tRNAs), 2 ribosomal RNAs (rRNAs), and 13 polypeptides that function exclusively for mitochondrial-related processes. The 13 polypeptides corresponding to respiratory chain subunits are: complex I subunits (7), complex III subunit (1), complex IV subunits (3), and complex V subunits (2)[2]. However, not all of the vital mitochondrial proteins are translated inside of the mitochondrion; only about 1% of mitochondrial proteins are synthesized within the mitochondrial matrix[24]. Mitochondrial proteins that are nuclear encoded and subsequently translated in the cytosol must be trafficked and integrated into the mitochondrion. Only precursor proteins containing a specialized signal sequence are granted entry across the mitochondrial membranes using specialized multimeric protein translocator complexes: translocase of the outer membrane (TOM), the translocase of the inner membrane (TIM), and the OXA complex.
Divergent from nuclear DNA, mtDNA is not packaged like chromatin; rather it is organized into structures known as nucleoids. Since mtDNA lacks the attributes embodied in chromatin, the nucleoid structure functions to provide sequential regulation of mtDNA related processes, while simultaneously protecting against degradation and damage[2]. Dependent on the cell line, nucleoids contain between one and eight mtDNA copies[7]. While it remains challenging to fully characterize the composition of a nucleoid, the protein TFAM (transcription factor A of mitochondria) functions as a critical component of packaging that non-selectively binds to mtDNA to create negative supercoiling, thereby significantly compacting the mitochondrial genome[2,22]. A unique attribute is their close association to the mitochondrial inner membrane, via anchoring, as visualized by high resolution microscopy[2].
Multiple copies of mtDNA can be contained within a single cell and are either identical (homoplasmy) or dissimilar (heteroplasmy)[2]. Some potential reasons mitochondrial genomes may become heteroplasmic are: maternal transmission, location of the mtDNA relative to oxidative phosphorylation, lack of histones, and the truncated efficiency of the mitochondrial DNA repair machinery[2]. While some mtDNA mutations may be considered pathogenic, there also exists a potential for others to have no effect. A pathogenic mutation can be overcome if the amount of non-mutated (wild-type) mtDNA is greater than mutated mtDNA within the same cell. While an advantage lies in this scenario, the opposite circumstance also exists and can induce deleterious effects. When the amount of mutated mtDNA is greater than non-mutated mtDNA, it will likely result in a mitochondrial-related disease. This phenomenon is known as the threshold effect and is a critical component regarding the existence of numerous mitochondrial myopathies[2]. Further, due to these varying ratios of normal to diseased mtDNA, its imbalance will likely cause a spectrum of mitochondrial defects leading to a variation in symptom severity per patient.
Mitochondrial diseases
Overview
Mitochondrial diseases are a collection of illnesses arising from defects relative to the respiratory chain and oxidative phosphorylation[25]. As of current, the United Mitochondrial Disease Foundation (UMDF) has compiled a comprehensive list of varying mitochondrial diseases [Table 2][26]. Mitochondrial diseases can either involve affecting a single organ or numerous organ systems, which often phenotypically have a neurologic and myopathic presentation[27]. These diseases have an ability to arise from either nuclear or mitochondrial DNA gene mutation(s), since mitochondria rely heavily on the interaction between these genomes[27]. As previously mentioned, the thirteen polypeptides mtDNA encodes for are respiratory chain subunits and, in addition, these complexes contain several subunits encoded by the nuclear genome[25]. Aside from their participation as integral members of oxidative phosphorylation, the gene products encoded by the nuclear genome dictate specific mitochondrial functions[25]. Mitochondrial diseases can participate in numerous genetic disorders and partake in the aging process[27].
Classified mitochondrial diseases according to the United Mitochondrial Disease Foundation[26]
| Mitochondrial diseases |
|---|
| Progressive infantile poliodystrophy (Alpers disease) |
| Autosomal dominant optic atrophy (ADOA) |
| Barth syndrome/lethal infantile cardiomyopathy (LIC) |
| Beta-oxidation defects |
| Carnitine-acyl-carnitine deficiency |
| Carnitine deficiency |
| Cerebral creatine deficiency syndromes (CCDS) |
| Co-enzyme Q10 deficiency |
| NADH dehydrogenase (NADH-CoQ reductase) deficiency (complex I deficiency) |
| Succinate dehydrogenase deficiency (complex II deficiency) |
| Ubiquinone-cytochrome c oxidoreductase deficiency (complex III deficiency) |
| Cytochrome c oxidase deficiency (complex IV deficiency/COX deficiency) |
| ATP synthase deficiency (complex V deficiency) |
| Chronic progressive external ophthalmoplegia syndrome (CPEO) |
| CPT I deficiency |
| CPT II deficiency |
| Kearns-Sayre syndrome (KSS) |
| Lactic acidosis |
| Leber’s hereditary optic neuropathy (LHON) |
| LBSL - leukodystrophy |
| Long-chain acyl-CoA dehydrongenase deficiency (LCAD) |
| LCHAD |
| Subacute necrotizing encephalomyelopathy (Leigh disease or syndrome) |
| Luft disease |
| Multiple acyl-CoA dehydrogenase deficiency (MAD/glutaric aciduria type II) |
| Medium-chain acyl-CoA dehydrongenase deficiency (MCAD) |
| Mitochondrial encephalomyopathy lactic acidosis and strokelike episodes (MELAS) |
| Myoclonic epilepsy and ragged-red fiber disease (MERRF) |
| Mitochondrial recessive ataxia syndrome (MIRAS) |
| Mitochondrial cytopathy |
| Mitochondrial DNA depletion |
| Mitochondrial encephalopathy |
| Myoneurogastointestinal disorder and encephalopathy (MNGIE) |
| Neuropathy, ataxia, and retinitis pigmentosa (NARP) |
| Pearson syndrome |
| Pyruvate carboxylase deficiency |
| Pyruvate dehydrogenase complex deficiency (PDCD/PDH) |
| POLG2 mutations |
| Short-chain acyl-CoA dehydrogenase deficiency (SCAD) |
| Encephalopathy and possibly liver disease or cardiomyopathy (SCHAD) |
| Very long-chain acyl-CoA dehydrongenase deficiency (VLCAD) |
Of these mutations, upwards of 250 pathogenic mutations and rearrangements have been identified in a multitude of diseases affecting both central nervous and muscle systems[28]. After collecting and analyzing epidemiological data regarding both childhood and adult mitochondrial diseases, Schaefer et al.[29] suggests the lowest prevalence of mitochondrial disease is approximately 1 in 5000. However, mitochondrial diseases could be far less obscure; accurate identification is limited by: disease onset related to patient age, the dual role and interplay of nuclear and mitochondrial genomes, symptoms similar to other diseases, as well as a spectrum of disease characteristics[30]. This begs the question that given the possible prevalence of mtDNA mutations, what differentiates an asymptomatic carrier, oligosymptomatic patient, and an individual presenting a phenotypic clinical manifestation of the disease? As mutant and wild-type mtDNA can exist in tandem, the clinical observance of an mtDNA pathogenic mutation is largely determined by the proportion of mutant to wild-type genomes in varying tissues[25]. Typically, greater percentages of mutant heteroplasmy are linked with a severe clinical presentation and an earlier disease onset[4]. The variability of mitochondrial heteroplasmy can impact the clinical phenotypic manifestations of disease. Generally, the mtDNA deletion phenotypic threshold value is approximately 60%, and for other mtDNA mutations is close to 90%[31,32]. The critical threshold level seems to vary within different tissues and organs, which is directly due to their energy demand[33]. The causal relationship between heteroplasmy and the threshold effect can explain clinical phenotype variations in one individual, or the same family of individuals, via their percentage of mutated mtDNA[27]. Moreover, on a global cellular level, mitotic segregation can further explain how some patients can possess a clinical phenotype in childhood and alternative phenotype in adulthood[25].
Mitochondrial diseases have been divided into the following categories: mtDNA mutations, nuclear DNA mutations, and intergenomic signaling defects[25]. The first category, diseases attributed to mtDNA, involves the identification and characterization of pathogenic mutations with a larger number of mtDNA deletions and duplications[25]. Conversely, the second category, nuclear DNA mutations, focuses on how the defects of the respiratory chain due to mendelian genetics comprises a considerable portion of mitochondrial diseases[25]. Lastly, the third category, intergenomic signaling defects, are mutations that arise from the nuclear genome that directly affect the mtDNA copy number and its stability[25]. Mitochondrial disease can present itself as either a distinct clinical syndrome or an myriad of disease phenotypes[27].
Diagnosing a disease of this variety can remain challenging as its fundamental roots may point to either mitochondrial or nuclear DNA, which can be determined using a two-tiered approach that includes both non-molecular (neuroimaging, muscle biopsy, cardiac and lactate concentration evaluation) and molecular (sequencing either nuclear or mitochondrial DNA for pathogenic mutations from isolated tissue) genetic testing methods[34]. The vast diversity of causes leading to mitochondrial encephalomyopathies correlates to a diverse patient treatment plan that relies on: physiological, biochemical, and genetic approaches[25]. Medicine, surgery, prosthetics, and perhaps dialysis or transfusions are at the forefront of physiological treatment options[25]. Whereas biochemical treatments rely on the supplementation of oxidative phosphorylation components, and the reduction of accompanying toxicity[25]. As a complement to the physiological and biochemical approaches, genetic treatments may depend on either genetic counseling, the alteration of heteroplasmy, and the cellular import of exogenous biological material (i.e. nucleic acids/proteins)[25].
MERRF and MELAS
Of particular interest, MERRF and MELAS are debilitating diseases that are among the multisystemic mitochondrial diseases/syndromes that are clinically defined, alongside Kearns-Sayre syndrome (KSS)[35]. MERRF and MELAS, both classified as orphan diseases, are caused by a mtDNA point mutation that is responsible for disrupting the pairing of the codon-anticodon that is necessary for protein synthesis by disturbing the tRNAs tri-dimensional structure, as well as any associated post-transcriptional modifications[2].
MERRF syndrome, commonly associated with an origination in childhood is described as a multisystem disease that is characterized by spontaneous muscle contractions, generalized epilepsy, loss of control regarding bodily movements, dementia and weakness[36]. Uncommon pathogenic variants within mtDNA encoded tRNA genes (defined as MT-TX) correlated with MERRF are: MT-TF, MT-TL1, MT-TI, and MT-TP[36]. However, the most prominent pathogenic variant (present in approximately 80% patients or greater) is MT-TK (encodes for tRNALys), which can be observed by an A-to-G nucleotide switch at position 8344 (m.8344A>G) [Figure 1][36]. While the clinically detectable pathogenic variants associated with MERRF can mostly be found in all tissues, the existence of heteroplasmy can cause mutated mtDNA to be variably distributed in tissues that leads to a changeable disease-triggering threshold level[36].
Similar to MERRF, MELAS is also a multisystem disease, with an origination usually in childhood, that is characterized by stroke-like episodes, dementia, seizures, lactic acidosis, ragged red fibers (visible during muscle biopsy), vomiting and headaches[37,38]. The most prominent pathogenic variant (present in approximately 80% patients) is MT-TL1 (encodes for tRNALeu(UUA/UUG)), which can be observed by an A-to-G nucleotide switch at position 3243 (m.3243A>G) [Figure 1][37]. Other common pathogenic variants, such as MT-ND5, are associated with MELAS[37]. Confirming a patient positive for MELAS focuses on correctly diagnosing phenotypic characteristics associated with the disease, as well as employing genetic testing for further validation (extraction of mtDNA from leukocytes acts as a reliable source)[37]. Like MERRF, the existence of mtDNA heteroplasmy can cause mutated mtDNA to be variously distributed in tissues, which leads to a mutable disease-triggering threshold level[37]. Studies have shown the A3243G mutation affects mitochondrial protein synthesis, as well as the respiratory chain, when the threshold of approximately 85% mutant mtDNA is reached (as determined by Kobayashi et al.[39]); even though mutant mtDNA levels vary among individuals, as well as organs and tissues of a single individual[40]. On average, individuals with MELAS do not have a favorable prognosis, as the 34.5 ± 19 years is the average age of death[38].
MERRF and MELAS - current advancements in gene therapy
The current state of care for MERRF and MELAS patients focuses on a combination of genetic counseling, surgery, and palliative treatment. Potential reasons why a pharmacological cure does not currently exist (for either disease) may be attributed to the location of mtDNA within the mitochondrial matrix, followed by an inability to correct the disease-causing point mutation. It seems plausible that developing a gold-standard capable of overcoming these hurdles, and cure a mitochondrial disease, is likely difficult at best. The field of gene therapy may offer hopeful and potentially realistic possibilities that may lie beyond routine patient symptom management. It is an emerging field that seeks to treat or prevent disease using viral and/or nonviral modalities by inducing correction (of a particular cellular gene) via an exogenous payload (i.e., nucleic acids). While gene therapy may theoretically sound like a promising prospective, downsides such as mistargeting of the exogenous payload, transient correction, inciting an immune response, and cellular apoptosis, do exist. While these existing downsides have a direct relationship with nuclear targeting, integrating mitochondrial biology brings forth an additional obstacle a gene therapy agent would have to be capable of overcoming.
For these reasons, previous gene therapy research efforts, with a focus on correcting MERRF and MELAS diseases, have been conservatively progressing for the past few decades. Gene therapy research efforts have been focused on three main approaches: nucleic acid delivery, peptide-mediated therapy, and cleavage of pathogenic mutations by mitochondrial-targeted transcription activator-like effector nucleases (mitoTALENS) [Table 3][41-56].
Corrective gene therapy approaches for MERRF and MELAS
| Gene therapy approach | Mitochondrial disease | |
|---|---|---|
| MERRF | MELAS | |
| Nucleic acid delivery | Kolesnikova et al.[43], 2004 Mahata et al.[41], 2005 Mahata et al.[42], 2006 | Karicheva et al.[44], 2011 |
| Peptide-mediated therapy | Chang et al.[50], 2013 Chang et al.[51], 2013 Muratovska et al.[53], 2001 Perli et al.[49], 2016 Taylor et al.[54], 1997 | Chang et al.[52], 2017 Li and Guan[47], 2010 Park et al.[46], 2008 Perli et al.[48], 2014 Perli et al.[49], 2016 Sasarman et al.[45], 2008 |
| mitoTALENs | Bacman et al.[55], 2015 Hashimoto et al.[56], 2015 | Bacman et al.[55], 2015 Hashimoto et al.[56], 2015 |
The goal of nucleic acid therapy is to deliver exogenous material to mitochondria harboring pathogenic mutations to restore function. Mahata et al.[41,42] demonstrated the RNA import complex from Leishmania can induce tRNALys import into mitoplasts isolated from a transmitochondrial cybrid MERRF cell line and in MERRF cybrid cells. Following the import of tRNALys, Mahata et al.[41,42] observed restored mitochondrial translation to near-wild-type levels and suppression of mutant polypeptide production. Likewise, Kolesnikova et al.[43] demonstrated that mitochondrial functions were partially restored due to the import of yeast tRNALys into homoplasmic transmitochondrial cybrid MERRF lines and primary human fibroblasts that were 70% ± 5% heteroplasmic for MERRF. Following this research, Karicheva et al.[44] transfected cybrid cells that were 90% ± 5% heteroplasmic for MELAS with tRNALeu(UUR) transcripts. Authors observed an improvement in mitochondrial translation, higher levels of mtDNA encoded respiratory subunits, and increased respiration activity[44].
Moreover, peptide-mediated therapy uses peptides to facilitate restoring mitochondrial function. Overexpression of mitochondrial translation elongation factors, namely EFTu or EFG2, results in the partial suppression of respiratory chain deficiency in MELAS myoblasts, but not in MERRF myoblasts[45]. Additionally, researchers determined that overexpression of leucyl-tRNA synthetase corrected mitochondrial dysfunction in human mitochondria carrying either the nearly homoplasmic A3243G or A83443G mutation[46-49]. Specifically, Perli et al.[48] demonstrated the carboxy-terminal domain of leucyl-tRNA synthetase is sufficient for cell rescue, and is even more efficient than the whole enzyme at doing so. Researchers were able to further isolate the rescuing activity of the carboxy-terminal domain of leucyl-tRNA synthetase; with this knowledge, they designed short peptide sequences capable of cell rescue that effectively bind with high affinity to both wild-type and mutant human mt-tRNALeu(UUR) and mt-tRNALys[49]. Chang et al.[50,51] conducted several studies suggesting that a Pep-1 peptide delivery system is capable of rescuing mitochondria containing the MERRF mutation. Pep-1, a cell-penetrating peptide, can translocate mtDNA from wild-type mitochondria into cybrid cell models of MERRF[50,51]. After three days of treatment with Pep-1-mediated mitochondrial delivery, mitochondrial function was recovered and cells were maintained for twenty-one days[50]. Recently, Chang et al.[52] demonstrated that Pep-1-mediated mitochondrial delivery can ameliorate mitochondrial function in cells containing the pathogenic MELAS mutation. Further, Muratovska et al.[53] and Taylor et al.[54] independently synthesized peptide nucleic acids (PNA) complementary to the sequence containing the MERRF mutation; upon entry into the mitochondria, PNA oligomers are able to selectively inhibit replication and translation of the mutant mtDNA template using in vitro and cell-based studies, respectively.
Lastly, mitochondrial-targeted transcription activator-like effector nucleases (mitoTALENS) are designed to cleave specific sequences of mtDNA that contain a pathogenic mutation[55,56]. When tested on heteroplasmic cybrid cells harboring MERRF and MELAS, respectively, mitoTALENS permanently reduced the mutant load and resulted in the return to normal mitochondrial respiratory activity[55,56].
Concluding remarks and future directions
A review of gene therapy strategies targeting the mtDNA disease’s, MERRF and MELAS, presents a series of realistic possibilities for their potential correction. Understanding the backstory of mitochondrial biology sets the tone for further understanding the path of mitochondrial dysfunction when considering an mtDNA-causing mitochondrial disease. While multiple forms of care exist for patients living with either MERRF or MELAS, none are capable of permanently correcting its associated mitochondrial mutation. Even if a certain approach is capable of restoring mitochondrial function, one must be prepared to answer several general arguments: (1) does the treatment approach lead to permanent or transient correction; (2) if the treatment was initially conducted in an in vitro setting, does it have the potential to be translated to an in vivo setting; and (3) if it can be adapted to an in vivo environment, what type of an immunogenicity is it met with? Rationally speaking, a researcher targeting MERRF or MELAS diseases must be both practiced and prudent when developing a gene therapy vector. Future directions surrounding research for these diseases should focus on developing a therapeutic vector that is capable of answering the aforementioned questions relating to: overcoming cellular and mitochondrial barriers, establish long-term (if not permanent) correction, and exhibiting a low (to negative) immunogenicity profile in patients.
Declarations
AcknowledgmentsThe authors take special acknowledgment to Ueli Gubler, Ph.D. for his scientific guidance and expertise regarding the subject matter.
Authors’ contributionsConcept and design: Agresti CA, Tolias P
Data acquisition and analysis: Agresti CA, Halkiadakis PN
Manuscript preparation: Agresti CA, Halkiadakis PN, Tolias P
Critical revision and finalizing of the manuscript: Agresti CA, Tolias P
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
2. Renard P, Michel S, Rommelaere G, Arnould T. Mammalian mitochodrial genetics, genomics and turnover In: Lou PH, Petersen N, editors. Cellular Bioenergetics in Health and Diseases: New Perspectives in Mitochondrial Biology. Kerala, India: Research Signpost; 2012. pp. 1-83.
4. Cohen B. Introduction: Mitochondrial Medicine. In: Saneto RP, Parikh S, Cohen BH, editors. Mitochondrial Case Studies: Underlying Mechanisms and Diagnosis. Elsevier; 2016. pp. 1-9.
5. Robin ED, Wong R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J Cell Physiol 1988;136:507-13.
6. Iwasa J, Marshall M. Karp's Cell and Molecular Biology. 8th ed. John Wiley & Sons, Inc.; 2016. pp. 168–97.
8. Westrate LM, Drocco JA, Martin KR, Hlavacek WS, MacKeigan JP. Mitochondrial morphological features are associated with fission and fusion events. PLoS One 2014;9:e95265.
9. Raulfs EC, O'Carroll IP, Dos Santos PC, Unciuleac MC, Dean DR. In vivo iron-sulfur cluster formation. Proc Natl Acad Sci U S A 2008;105:8591-6.
11. Chiabrando D, Mercurio S, Tolosano E. Heme and erythropoiesis: more than a structural role. Haematologica 2014;99:973-83.
12. Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res 2011;52:2111-35.
13. Ah Mew N, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML. Urea Cycle Disorders Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. SourceGeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2003.
14. Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli C, Patergnani S, Poletti F, Rimessi A, Suski JM, Wieckowski MR, Pinton P. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012;12:22-85.
15. Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 2000;29:222-30.
16. Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Rep 2011;12:901-10.
17. Kuwabara M, Asanuma T, Niwa K, Inanami O. Regulation of cell survival and death signals induced by oxidative stress. J Clin Biochem Nutr 2008;43:51-7.
18. Nass MM, Nass S. Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions. J Cell Biol 1963;19:593-611.
19. Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature 1981;290:457-65.
20. Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 1999;23:147.
21. Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1999;1410:103-23.
22. Nicholls TJ, Minczuk M. In D-loop: 40 years of mitochondrial 7S DNA. Exp Gerontol 2014;56:175-81.
23. Kuhl I, Miranda M, Posse V, Milenkovic D, Mourier A, Siira SJ, Bonekamp NA, Neumann U, Filipovska A, Polosa PL, Gustafsson CM, Larsson NG. POLRMT regulates the switch between replication primer formation and gene expression of mammalian mtDNA. Sci Adv 2016;2:e1600963.
24. Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell 2009;138:628-44.
25. Schon EA, DiMauro S. Medicinal and genetic approaches to the treatment of mitochondrial disease. Curr Med Chem 2003;10:2523-33.
26. United Mitochodnrial Disease Foundation. Types of Mitochondrial Diseases. Available from: http://www.umdf.org/types/ [Last access on 28 Jun 2018].
27. Chinnery PF. Mitochondrial Disorders Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2000.
29. Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders--past, present and future. Biochim Biophys Acta 2004;1659:115-20.
30. Saneto RP, Sedensky MM. Mitochondrial disease in childhood: mtDNA encoded. Neurotherapeutics 2013;10:199-211.
31. Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J 2003;370:751-62.
32. Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990;61:931-7.
35. Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M, Simonetti S, Angelini C, Donati MA, Garcia C, Martinuzzi A, Mosewich R, Servidei S, Zammarchi E, Bonilla E, DeVivo DC, Rowland LP, Schon EA, DiMauro S. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol 1992;31:391-8.
36. DiMauro S, Hirano M. MERRF. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2003.
37. DiMauro S, Hirano M. MELAS. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2001.
38. Marin S, Haas R. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). In: Saneto RP, Parikh S, Cohen BH, editors. Mitochondrial Case Studies: Underlying Mechanisms of Disease. Elsevier; 2016. pp. 13-29.
39. Kobayashi Y, Momoi MY, Tominaga K, Shimoizumi H, Nihei K, Yanagisawa M, Kagawa Y, Ohta S. Respiration-deficient cells are caused by a single point mutation in the mitochondrial tRNA-Leu (UUR) gene in mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS). Am J Hum Genet 1991;49:590-9.
40. Mehrazin M, Shanske S, Kaufmann P, Wei Y, Coku J, Engelstad K, Naini A, De Vivo DC, DiMauro S. Longitudinal Changes of mtDNA A3243G Mutation Load and Level of Functioning in MELAS. Am J Med Genet A 2009;149A:584-7.
41. Mahata B, Bhattacharyya SN, Mukherjee S, Adhya S. Correction of translational defects in patient-derived mutant mitochondria by complex-mediated import of a cytoplasmic tRNA. J Biol Chem 2005;280:5141-4.
42. Mahata B, Mukherjee S, Mishra S, Bandyopadhyay A, Adhya S. Functional delivery of a cytosolic tRNA into mutant mitochondria of human cells. Science 2006;314:471-4.
43. Kolesnikova OA, Entelis NS, Jacquin-Becker C, Goltzene F, Chrzanowska-Lightowlers ZM, Lightowlers RN, Martin RP, Tarassov I. Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum Mol Genet 2004;13:2519-34.
44. Karicheva OZ, Kolesnikova OA, Schirtz T, Vysokikh MY, Mager-Heckel AM, Lombes A, Boucheham A, Krasheninnikov IA, Martin RP, Entelis N, Tarassov I. Correction of the consequences of mitochondrial 3243A>G mutation in the MT-TL1 gene causing the MELAS syndrome by tRNA import into mitochondria. Nucleic Acids Res 2011;39:8173-86.
45. Sasarman F, Antonicka H, Shoubridge EA. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum Mol Genet 2008;17:3697-707.
46. Park H, Davidson E, King MP. Overexpressed mitochondrial leucyl-tRNA synthetase suppresses the A3243G mutation in the mitochondrial tRNA(Leu(UUR)) gene. RNA 2008;14:2407-16.
47. Li R, Guan MX. Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol Cell Biol 2010;30:2147-54.
48. Perli E, Giordano C, Pisano A, Montanari A, Campese AF, Reyes A, Ghezzi D, Nasca A, Tuppen HA, Orlandi M, Di Micco P, Poser E, Taylor RW, Colotti G, Francisci S, Morea V, Frontali L, Zeviani M, d'Amati G. The isolated carboxy-terminal domain of human mitochondrial leucyl-tRNA synthetase rescues the pathological phenotype of mitochondrial tRNA mutations in human cells. EMBO Mol Med 2014;6:169-82.
49. Perli E, Fiorillo A, Giordano C, Pisano A, Montanari A, Grazioli P, Campese AF, Di Micco P, Tuppen HA, Genovese I, Poser E, Preziuso C, Taylor RW, Morea V, Colotti G, d'Amati G. Short peptides from leucyl-tRNA synthetase rescue disease-causing mitochondrial tRNA point mutations. Hum Mol Genet 2016;25:903-15.
50. Chang JC, Liu KH, Chuang CS, Su HL, Wei YH, Kuo SJ, Liu CS. Treatment of human cells derived from MERRF syndrome by peptide-mediated mitochondrial delivery. Cytotherapy 2013;15:1580-96.
51. Chang JC, Liu KH, Li YC, Kou SJ, Wei YH, Chuang CS, Hsieh M, Liu CS. Functional recovery of human cells harbouring the mitochondrial DNA mutation MERRF A8344G via peptide-mediated mitochondrial delivery. Neurosignals 2013;21:160-73.
52. Chang JC, Hoel F, Liu KH, Wei YH, Cheng FC, Kuo SJ, Tronstad KJ, Liu CS. Peptide-mediated delivery of donor mitochondria improves mitochondrial function and cell viability in human cybrid cells with the MELAS A3243G mutation. Sci Rep 2017;7:10710.
53. Muratovska A, Lightowlers RN, Taylor RW, Turnbull DM, Smith RA, Wilce JA, Martin SW, Murphy MP. Targeting peptide nucleic acid (PNA) oligomers to mitochondria within cells by conjugation to lipophilic cations: implications for mitochondrial DNA replication, expression and disease. Nucleic Acids Res 2001;29:1852-63.
54. Taylor RW, Chinnery PF, Turnbull DM, Lightowlers RN. Selective inhibition of mutant human mitochondrial DNA replication in vitro by peptide nucleic acids. Nat Genet 1997;15:212-5.
55. Bacman SR, Hashimoto M, Peralta S, Falk MJ, Chomyn A, Chan DC, Williams SL, Moraes CT. mitoTALENs as DNA editing tools for mitochondrial diseases. Mitochondrion 2015;24:S22.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Agresti CA, Halkiadakis PN, Tolias P. MERRF and MELAS: current gene therapy trends and approaches. J Transl Genet Genom 2018;2:9. http://dx.doi.org/10.20517/jtgg.2018.05
AMA Style
Agresti CA, Halkiadakis PN, Tolias P. MERRF and MELAS: current gene therapy trends and approaches. Journal of Translational Genetics and Genomics. 2018; 2: 9. http://dx.doi.org/10.20517/jtgg.2018.05
Chicago/Turabian Style
Agresti, Ciara Ann, Penelope Nicole Halkiadakis, Peter Tolias. 2018. "MERRF and MELAS: current gene therapy trends and approaches" Journal of Translational Genetics and Genomics. 2: 9. http://dx.doi.org/10.20517/jtgg.2018.05
ACS Style
Agresti, CA.; Halkiadakis PN.; Tolias P. MERRF and MELAS: current gene therapy trends and approaches. J. Transl. Genet. Genom. 2018, 2, 9. http://dx.doi.org/10.20517/jtgg.2018.05
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 30 clicks
Cite This Article 30 clicks

 Like This Article 42
likes
Like This Article 42
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.