
Exome sequencing identifies FLNC and ADD3 variants in a family with cardiomyopathy
 0
0
Abstract
Aim: Idiopathic cardiomyopathy is often genetic in origin, typically autosomal dominant, and restrictive cardiomyopathy (RCM) is the rarest form. Clinically, RCM prognosis is poor with most patients requiring heart transplant due to impaired diastolic function leading to heart failure. In some cases, desminopathy is also observed, whereby desmin protein aggregates in the myocardium. Many genes are known to be involved in cardiomyopathy, and we sought to find the pathogenic mutation of a four-generation family with RCM and desminopathy.
Methods: We employed whole exome sequence analysis of four RCM patients from the family, to identify the underlying pathogenic mutation(s).
Results: Analysis of the exome data led to identification of two putative pathogenic variants. One, a nonsense mutation in ADD3, encoding adducin 3, was found in the proband and his affected daughter, but not other family members. The other was a missense mutation (G1546S) in the gene encoding filamin C (FLNC), found in all of the affected individuals. Both of these proteins interact with proteins involved in sarcomere function, and are expressed in both heart and skeletal muscle. FLNC has recently been implicated as a cardiomyopathy gene, but ADD3 has not at this point.
Conclusion: We present bioinformatic analyses that suggest that the FLNC variant is the major pathogenic variant affecting this family, but cannot rule out some contribution of the ADD3 mutation in at least two patients.
Keywords
Introduction
Restrictive cardiomyopathy (RCM) is a cardiac disease in which the diastolic function of the heart is impaired by increased myocardial stiffness, decreased ventricular filling and biatrial enlargement[1-3]. Prognosis is very poor, often requiring cardiac transplantation as a result of heart failure. RCM is the rarest of the classical types of cardiomyopathy, accounting for approximately 5% of all primary cardiomyopathy cases[4-6]. The other primary cardiomyopathies are classified on a morphological basis as hypertrophic (HCM), dilated (DCM), and arrhythmogenic right ventricular (ARVCM). In RCM, there is diastolic dysfunction but the overall size of the heart is typically normal, with minor enlargement of atria. Ventricular enlargement and other features can also occur, overlapping with features of the other primary cardiomyopathies. This phenotypic overlap demonstrates the clinical complexity of cardiomyopathy, and the variable expressivity from well over 1000 mutations in over 100 known cardiomyopathy genes[6]. Typically, these genes encode proteins of the sarcomere, z-disk domain, intermediate filaments, ion channel, intercalated disk, and cytoskeletal proteins[3,7-10]. This variable expressivity in cardiomyopathy is seen even within the same family, with multiple diagnoses (including this family and other examples)[7,9,11,12]. RCM can also occur from infiltrative processes, such as amyloid or desmin deposition, often due to genetic mutations. Although a few cases of recessive inheritance in RCM have been described, most RCM pedigrees best fit autosomal dominant inheritance, consistent with the other types of cardiomyopathy[13-15]. Such gene mutations can occur de novo as well, with no prior family history. Mutations associated with RCM have been found in MYH7, TNNI3, TNNT2, ACTC, BAG3, DES, MYL2, MYL3, MYPN, TPMI, TTN, and TTR[16]. More recently, the genes FLNC, TNNC1, and MYBPC3 have also been implicated[13,17,18].
In 2001, our group described a four-generation family presenting with RCM and desminopathy, but not skeletal myopathy. Known cardiomyopathy-causing genes were eliminated at the time, and the family was subsequently studied in low-density linkage analysis using 430 microsatellite markers across the genome. The study assumed an autosomal dominant model as ascertained from the pedigree. The most promising locus was at chromosome 10q23, reaching significance (LOD score 4.0, 1:10,000 odds in favor of linkage) for two-point haplotype analysis, but the overall linkage was weak because no single polymorphisms met the minimum LOD of 3.0[19]. Putative candidate gene analysis at the time was prohibitive because the linked interval was very large and contained over 40 genes (none of which had been implicated in cardiomyopathy). In addition, it was among the last of the genomic regions characterized in the Human Genome project, with the sequence continuing to be updated even after 2005. Thus, a genome-wide exome sequencing (also known as whole exome sequencing, WES) analysis was undertaken in four affected individuals from the family, not only to provide sequence of the genes in or near the linked interval, but to also analyze all other protein-coding genes in an unbiased fashion, since the linkage data was not very strong given the size of the family. Using this approach, we identified two variants of interest. One, a novel nonsense mutation in ADD3, was in the linked interval on chromosome 10 but was only present in the proband and his affected child; the other was a missense mutation on chromosome 7, in the filamin C gene (FLNC), which appears to be the major pathogenic variant in this family, as described here.
Methods
Pedigree and subject clinical data
Cardiac phenotype and family structure information was presented previously, in which affected individuals with available medical records had a condition consistent with diagnosis of RCM, with one individual also having left ventricular hypertrophy, overlapping HCM phenotype[19,20]. In this report, an additional affected family member, a daughter of the proband, is included [Figure 1], and a table of clinical data is shown as well [Table 1]. The proband (code 141) had RCM with no ventricular hypertrophy and received a heart transplant prior to age 10. The proband’s daughter’s cardiac condition best fit a diagnosis of HCM, rather than RCM, and she underwent cardiac transplantation prior to the age of 5 years, the youngest of any family member to need a transplant. Family member 139 was reported to have features of RCM and HCM.
Cardiac data from affected individuals in this study at time of first presentation
| ID | Age at presentation^ (years) | Exertional dyspnea | Hypertension | Anasarca | ECG: atrial flutter/ fibrillation | ECG: ST/T changes in left side leads | Echo*: left atrium changes | Echo*: right atrium changes | Echo*: left ventricle systolic function | Echo*: left ventricle hypertrophy | Right ventricular conduction delay |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 136 | 0-5 | Yes | Yes | No | No | Yes | Dilated | Dilated | Dilated | Normal | Yes |
| 137 | 35-40 | Yes | Yes | No | Yes | Yes | Dilated | Dilated | Normal | Normal | No |
| 138 | 20-25 | Yes | No | No | No | Yes | Dilated | Dilated | Normal | Normal | No |
| 139+ | 30-35 | Yes | No | No | No | Yes | Dilated | Dilated | Normal | Normal | No |
| 140 | 30-35 | No | No | No | No | Yes | Dilated | Normal | Normal | Normal | No |
| 141 | 5-10 | Yes | Yes | No | No | No | Dilated | Dilated | Normal | Normal | Yes |
| 147 | 30-35 | No | No | No | Yes | Yes | NA | NA | NA | NA | No |
| 148 | 25-30 | Yes | Yes | No | Yes | Yes | Dilated | Dilated | Normal | Normal | No |
| 150 | 10-15 | No | No | No | No | Yes | Dilated | Normal | Normal | Normal | No |
| 206 | 25-30 | Yes | Yes | Yes | No | Yes | Dilated | Dilated | Normal | Mild | Yes |
| 416 | 10-15 | No | No | No | No | No | Normal | Normal | Normal | Normal | Yes |
| 507 | 50-55 | Yes | Yes | No | Yes | Yes | Dilated | Dilated | Normal | Mild | No |
| 1661 | 0-5 | Yes | No | No | No | Yes | Dilated | Dilated | 70%# | Moderate | No |
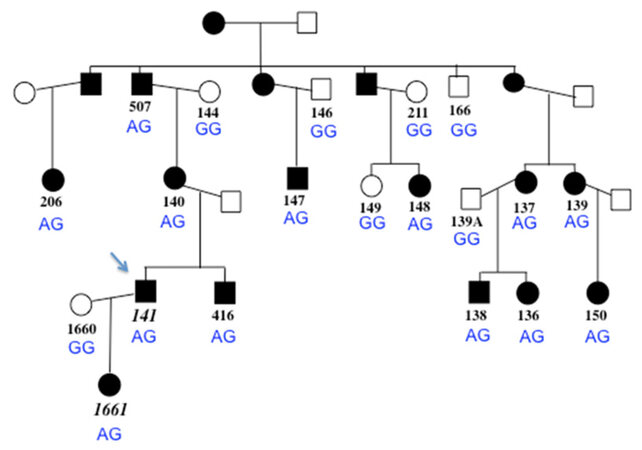
Figure 1. Pedigree of the family, with proband indicated with arrow. Black symbols represent individuals who are clinically affected with cardiomyopathy. Open shading indicates individuals who do not have features of cardiomyopathy. Subject numbers in italics (lower left) indicate the two individuals who are heterozygous for the ADD3 variant c.169C>T R57X (all other family members have wild type genotype, CC). The FLNC genotype at c.4636G>A G1546S is shown for each family member in blue; the A allele segregates with the clinical condition
Exome data generation
Family members were consented for this study per approved IRB protocol, and DNA was extracted from blood samples as previously reported[20]. Genomic DNA samples from the proband and three other affected members (proband’s mother and two first-cousins-once-removed) were prepared for exome sequencing using the Kapa Biosystems library preparation kit and 8 cycles of PCR amplification. WES employed the Illumina TruSeq exome kit v.1 (62.2 megabases, 20,794 genes, 201,121 exons) with the resulting enriched library subsequently sequenced on an Illlumina HiSeq 2000 machine at the Center for Pediatric Genomic Medicine, Children’s Mercy Hospital, Kansas City, to an average read depth of 80X.
Bioinformatic analysis of exome sequence reads
Alignment of reads with the human reference genome sequence NCBI v37 utilized the Genomic Short-read Nucleotide Alignment Program. Variants were called using the Genome Analysis Toolkit. In-house software, called Rapid Understanding of Nucleotide variant Effect Software, provided variant annotation, including: dbSNP entry number for previously-reported variants, Blocks Substitution Matrix (BLOSUM) scores for predicting whether missense changes are damaging to protein function, Human Gene Mutation Database annotation for previously reported mutations considered pathogenic, heterozygosity for the variant, variant type, and chromosomal location including name of the gene the variant lies within (if any). These data were first sorted to identify novel, putatively pathogenic variants in the proband’s sequence for an initial view of potential hits in the linked chromosome 10 region, which led to the identification of the ADD3 variant. Because this variant was not present in the other exomes, all four sets of exome data were filtered using custom Python scripts to screen the combined data for heterozygous variants common to the four affected subjects, with a minor allele frequency less than 1% based on data in dbSNP and within the Center’s own cumulative exome data. Candidate variants were further scrutinized by analysis of literature and database reviews on the functions of the host genes, to prioritize those genes with expression in cardiac muscle, previously reported in association with genetic basis of cardiomyopathy, or other evidence of putative involvement in sarcomere function. For example, the dbSTRING database was employed to find evidence of known interactions that would implicate cardiac function involvement. Variants (regardless of type) that appeared novel or very rare (allele frequency less than 1%) were prioritized after the filtering, with further prioritization based on any pathogenicity information from BLOSUM analysis. Candidates were also screened for presence in the Exome Aggregation Consortium database (exAC: http://exac.broadinstitute.org), gnomAD (gnomad.broadinstitute.org), NHLBI Exome Sequencing Project database (http://evs.gs.washington.edu/EVS/), and NHLBI whole genome sequencing project (TOPMED, https://bravo.sph.umich.edu/freeze5/hg38/) for further evidence as to whether these were previously reported and could be neutral polymorphisms. Multialignments obtained by the online ClustalOmega tool (http://www.ebi.ac.uk/Tools/msa/clustalo/) were also used to screen for conservation of candidate missense variants through evolution, and further pathogenicity prediction employed online software packages SNPs&GO (http://snps.biofold.org/snps-and-go/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), and Protein Variation Effect Analyzer (PROVEAN)(http://provean.jcvi.org/index.php).
Mutation validation
PCR oligonucleotide primers were designed to flank the exons containing the candidate variants using NCBI’s PrimerBLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). For the ADD3 variant, the PCR primers (producing a 151 bp product) were: reverse, 5’- TCCTCCTCCACCCCTCTTTTA and forward, 5’- ACATGATGGAGCAGAGGcAA with “c” designating a base change designed to create a BsrDI site only in the presence of the mutant allele (T), for a restriction digest-based system for a fast assay to genotype the other family members (mutant allele PCR product cut into two fragments of 25 bp and 126 bp). For validation and further analysis of the FLNC mutation, the primers were: forward, 5’- GCTCCTCAAGCCCCTTCAAGA; reverse, 5’- CTGAGGGAGAAGCAGAGGTCA, creating a PCR product of 241 bp. PCR primers to amplify and validate the additional candidate variants that were later ruled out as the main pathogenic mutation in this family were: BAG3 (forward 5’- ATTTGACCAAAGAGCTGCTGG, reverse 5’- GCTGGAGTTCATAGACCTGGAC), RBM20 (forward 5’- GGAGAAGTACCCGAGATCTGG, reverse 5’- CTGGCCTCGTCTTTCCTCC), MYOF (forward 5’-TAGGACCATCTACCTCCACTGT, reverse 5’- AGCCAGTTGTTACCCTGACTTC ), DNMBP (forward 5’- CAGTCAGAAGAACTCCGGGC, reverse 5’- CATTGTCAATCAGCCAGCGG), LDB3 (forward 5’-CCTGTTAGACAGGGGAATGGT, reverse 5’- TTAAGGATTTGGGCTGAAAGG), and XIRP1 (forward 5’- GGCAGCTGGCTGGGAGT, reverse 5’- AGCCATGAGAACCGTGACTG ). From subject DNA, PCR amplifications were performed using the Qiagen HotStar system (Germantown, MD, USA). All candidate variants were initially validated by Sanger sequencing of the PCR products (BigDye V3.1 cycle sequencing kit, ABI/ThermoFisher, Foster City, CA, USA) at the University of Florida’s Interdisciplinary Center for Biotechnology Research sequencing core, and genotyping of other family members to analyze variant segregation was accomplished by restriction digestion (ADD3) and Sanger DNA sequencing of both loci as described above.
Results
Over 100,000 variants were identified in each subject, representing loci where the subject was heterozygous or homozygous for the non-reference allele of known SNPs, or for a previously-unannotated or novel variant. After bioinformatic analysis of the proband’s data first, the variant of highest interest was a nonsense mutation in ADD3 (NM_011903.3, c.169C>T Arg57X, Figure 1), due to its being novel, clearly disruptive, within the linked interval, and the fact that its encoded protein, gamma adducin, is involved in actin/spectrin assembly.
To validate this variant, we first designed a PCR-RFLP test by altering one nearby base in one of the PCR primers. However, restriction digestion patterns were faint and difficult to resolve [Figure 2A], and thus DNA sequencing of this region (amplified with more distant primers) was performed to genotype the family [Figure 2B]. Surprisingly, this mutation was only present in the proband and his affected daughter (both heterozygous) but no other family members. We concluded that this mutation was not the main causative mutation in the family, but could potentially have functional impact given predicted haploinsufficiency at this locus.
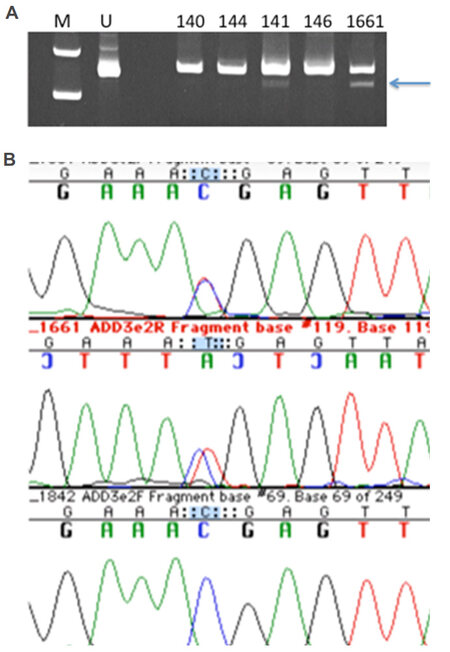
Figure 2. Panel A shows photograph of ethidium bromide-stained polyacrylamide gel of the BspDI digests to validate and genotype the ADD3 mutation c.169C>T Arg57X. M is the molecular weight marker lane, U contains uncut PCR product. The arrow indicates the smaller fragment validating the mutation in the proband 141 and his daughter 1661 and not in three other family members. Panel B shows the DNA sequencing chromatogram demonstrating the ADD3 heterozygous mutation in the proband's daughter's DNA [red (T)/blue (C) double peaks in top two lines; control sequence on the bottom line]
The other candidate variants were prioritized for further analysis by mutation type (frameshift, nonsense and missense mutations of greatest interest), predicted pathogenicity, gene expression in cardiac muscle (as evidenced in expression data at genecards.org), and function that was known or could feasibly be considered to be involved in heart structure or function. The top variants that were validated by sequencing and further considered (some in genes previously implicated in cardiomyopathy), but were ruled out because they did not segregate with the condition in the family, were: BAG3 rs199682693 missense (Arg473Gly), RBM20 rs1417635 missense (Trp768Ser), MYOF rs79732285 (splice donor variant), DNMBP rs769805859 c.3907G>T (Asp1303Tyr), LDB3 (also called ZASP, 5 bp del in a polypyrimidine tract in intron c.756-10 to -15), and XIRP1 rs138653994 (Gln1832Arg).
DNA sequencing was used to validate the variant at FLNC (NM_001127487.1) c.4636G>A G1546S variant [Figure 3], and this mutation was found to segregate with the condition in the family through multiple informative meioses. Based on the correct segregation pattern, this variant was further evaluated to assess factors regarding the probability of it being the pathogenic mutation in the family. This included the frequency of the FLNC c.4636G>A G1546S variant in the general population, location in conserved domains, and software-based prediction about functional impact, as detailed below.
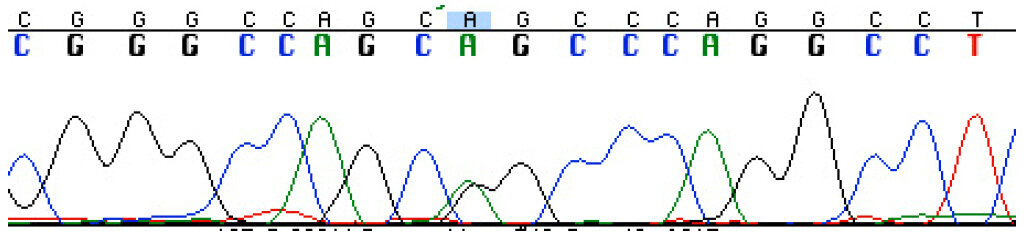
Figure 3. DNA sequencing chromatogram showing the FLNC heterozygous mutation c.4636G>A G1546S in the proband's DNA [green (A)/black(G) double peak under the blue box]
FLNC c.4636G>A G1546S was novel at first discovery, but more recently was entered in the NCBI dbSNP database (rs774263134) with extremely low MAF of 0.00002 (three heterozygotes in TOPMED (out of 62,784 genomes), no phenotype data available). dbSNP indicates that ExAc has found that allele once (frequency 0.000008) but it is not listed in the ExAC database when searched directly for FLNC. This variant is not reported in gnomAD or the NHLBI Exome Sequencing Project database. Thus, it is very rare, consistent with autosomal dominant pathogenicity for a rare disorder, but the presence of the allele in at least one database suggests that it could just be a neutral polymorphism. Since many of the exomes and genomes in the databases thus far come from studies of people suspected of having genetic conditions, it is possible that the heterozygous individuals (for example, in TOPMED, funded by NHLBI) do have heart-related conditions.
Next, predicted pathogenicity of c.4636G>A G1546S was analyzed using multiple software packages. FLNC residue 1546 is in the 14th (out of 24) Ig-like domain of filamin C, close to a recently identified RCM missense mutation S1624L17 and an HCM-related mutation, A1539T25. The Clustal alignment of this 14th domain peptide sequence, containing c.4636G>A G1546S, showed perfect conservation at the Gly 1546 residue among phylogenetically diverse organisms and filamin C isoforms [Figure 4]. The variant was then found to be deleterious in three programs based on algorithms considering biochemical change of the residue, conservation via multialignments, and 3D structural change. PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) found c.4636G>A G1546S to be “probably damaging”. This prediction used two different datasets of known damaging variants, HumDiv and HumVar, to compare the query variant with similar polymorphisms. Additionally, the PolyPhen-2 algorithm used sequences from UniProt, and 3D structural models from the Protein Data Bank to investigate evolutionary importance and potential disturbance of any binding sites. PROVEAN (http://provean.jcvi.org/index.php) computationally aligned 260 sequences, and predicted the FLNC variant to be “deleterious”. Finally, SNPs&GO (http://snps.biofold.org) used sequence alignments, 3D structure and function analyses to generate a prediction of “disease-causing”.

Figure 4. Clustal Omega alignment of filamin C protein sequences (3rd line and below) (residues 1500 to 1559) among phylogenetically diverse organisms, and aligned to human filamin A and filamin B (first two lines). The red box shows that the glycine at residue 1546 is completely conserved among species and in the human protein family. NCBI accession numbers are on the left, with species indicator. FICAL is flycatcher bird; PANTR is Pan troglodytes (chimpanzee); CANLF is Canis familiaris (dog). Asterisks below the residue indicate complete conservation. Colons below the residue indicate differences when compared with human filamin A and/or B
Discussion
Filamin C is a dimeric actin-cross-linking protein highly expressed in both skeletal and cardiac muscle, localized to the sarcolemma, Z-disk, and myotendinous junctions, and is associated with structural stability, mechanosensation, and intracellular signaling[21-23]. Recently, filamin C was also shown to mediate fast repair of myofibrillar microdamage in cardiomyocytes, in addition to Z-disk assembly[24]. Filamin C predominantly consists of 24 immunoglobulin-like domains spanning over 80% of the protein, with an actin-binding domain at the amino terminus and a dimerization domain at the carboxy terminus. The Ig-like domain region has been found to interact with over 90 proteins[24]. Initially, dominant disruptive mutations in FLNC were found in association with skeletal muscle phenotypes (OMIM no. 614065). More recently, FLNC mutations have been found in cardiac-only phenotypes, including HCM, DCM, RCM, and ARVCM[17,24-29]. The mutation described here, c.4636G>A G1546S, is located in the 14th Ig-like domain, which is thought to dimerize with the 15th domain just before the hinge region. Interference in the ability of these two domains to dimerize has been hypothesized to destabilize protein folding of the entire filamin C protein[22]. As a highly dynamic protein which interacts with many substrates, missense alterations in FLNC could feasibly destabilize interactions with other proteins and alter cytoplasmic distribution in cardiomyocytes[17,30,31]. The rarity of c.4636G>A G1546S, its bioinformatic analyses (including complete conservation of this residue through phylogenetically diverse organisms, and designation of predicted deleterious), its co-segregation with disease in the family, and reported FLNC mutations in HCM and RCM, together support the hypothesis that this is the pathogenic mutation in this family. This is the first large pedigree described with RCM and HCM bearing an FLNC mutation. Examination of this gene region in the original linkage data showed no strong evidence of linkage, however by chance none of the microsatellite markers available in that panel were near FLNC. Thus the moderate linkage to chromosome 10 appears to be coincidence, although ADD3 is in that region.
Abnormal cardiac tissue aggregate including filamin C protein was seen in a recently reported FLNC mutation case affecting only the heart, although not addressed in other FLNC mutation studies[17]. This has been termed “filaminopathy” similar to the situation in “desminopathy”. While that term now applies to a condition with abnormal tissue deposits in patients with DES mutations, including mutant desmin protein (e.g., Maerkens et al.[32], 2013), in early studies it was a descriptive term indicating presence and/or dominance of desmin in the tissue aggregate, without knowledge of underlying genetic cause. In the family described here, the desmin gene sequence is normal[19], yet cardiac aggregates containing desmin were seen. It is possible that mutant filamin C is also present and is precipitating the aggregation phenotype, which could attract other proteins to the complex as well as bystanders. This would be consistent with the Maerkens et al.[32] (2013) that filamin C (presumably normal protein sequence) was found in abundance in desminopathy deposits, along with a multitude of other proteins that appear to have also been trapped. It may be the gradual accumulation of these aggregates that are the underlying cause of cardiac muscle tissue failure, with such aggregates only forming in the presence of an abnormal protein, as seen in cardiac amyloidosis associated with missense mutations in the transthyretin (TTR) gene[33]. Although transthyretin deposits are extracellular and desmin deposits intracellular, both can stress cardiomyocytes. Such cardiomyopathy cases would fall within the realm of pathogenesis via toxic effect of misfolded protein accumulation, among the growing category of protein misfolding diseases, and could potentially benefit from therapies being developed for this molecular phenotype.
It is unclear why some FLNC mutations affect only skeletal muscle while others affect the heart (separately or in addition to skeletal muscle). It is possible that it may be related to the location or type of FLNC mutation. Among the reports of FLNC-related cardiomyopathy where the patients were ascertained due to heart phenotype, there was only one report of patients also having symptoms of skeletal myopathy or detection of protein deposits in skeletal muscle[30]. The predominantly-cardiac phenotype mutations include missense substitutions and truncating mutations are in functional domains scattered across the gene; the truncating mutations are thought to exert pathogenesis through haploinsufficiency rather than protein aggregation[24]. In patients with predominantly (or only) skeletal muscle myopathy, the mutations reported (A193T, M251T, in-frame p.Val930_Thr933del, F1720LfsX63, T2419M, and W2710X), are in regions and functional domains distinct from the cardiac-only reports, thus likely affecting the protein function differently such that cardiac muscle is preferentially spared[34,35].
There is no current evidence to support the contribution of the heterozygous ADD3 loss-of-function mutation to the phenotype, although this gene is down-regulated in dilated cardiomyopathy in the left ventricle compared to the right ventricle[36]. This would be consistent with loss of some ADD3 expression being contributory to a cardiomyopathy phenotype, as a modifier locus. Adducin 3 it is known to interact with actin, and also with ubiquitin C and ubiquitin-conjugating enzyme E2I (which also interacts with desmin) (BioGRID analysis, thebiogrid.org). The proband and his daughter were the youngest in this family to require heart transplants, with the exception of one of his 2nd cousins who needed transplant prior to age 10. The other living affected family members have not yet required transplantation, or did not need a transplant until later adulthood. This supports the possibility that this novel ADD3 mutation is contributing to the earlier onset of RCM in the proband and daughter. However, it could also be a neutral variant. It was not inherited from the proband’s mother, suggesting that it was either inherited from his father (DNA unavailable, but reported clinically unaffected) or arose de novo in one of the proband’s parents’ germ cells. There are no other clinical features that are specific to the proband and his daughter. The other candidate variants initially examined are unlikely to be contributing to the phenotype because they are not as rare (present in dbSNP) and/or were predicted to have lower pathogenic potential.
The power and efficiency of WES is demonstrated in the identification of the FLNC mutation in this family, and other pathogenic genes in other RCM pedigrees[8,11,28], dilated cardiomyopathy[37], and in disease-gene discovery in general[38]. Although there are commercially available panels to test for mutations in known cardiomyopathy genes, WES is an unbiased search for such mutations and has the power to identify new cardiomyopathy genes. However, the disadvantage is that many variants have to be filtered and validated, to identify the likely candidate, with the risk of missing a pathogenic variant if filtering choices are not appropriate.
Declarations
AcknowledgmentsThe authors wish to thank the subjects, the scientists and the physicians who contributed to the previous study of this family setting the stage for this report: Anil Kumar, M.D., Jun Zhang, Ph.D., Lee Kaplan, Ph.D., Michelle Burch, M.S., Angela Hadjipanayis, Ph.D., Julie Bokor, Ph.D., Heather Stalker, M.S., and Melissa Maisenbacher, M.S. We also acknowledge the help of the NIH NHGRI Medical Sequencing Project in sequencing exons of some candidate genes in the linked region. We thank Darrell Dinwiddie, Ph.D., and Emily Farrow, Ph.D. for technical expertise in the exome data generation at the Center for Pediatric Genomic Medicine, Children’s Mercy Hospital, Kansas City, and Dr. Alberto Riva for helping the first author with Python script.
Authors’ contributionsExome data validation experiments, bioinformatics, analysis of FLNC variant within the family, and manuscript preparation: Sanoja AJ
Exome data validation experiments and analysis of ADD3 variant within the family, manuscript review: Li H Clinical data and interpretation of phenotype differences, manuscript review: Fricker FJ
Provided exome data, manuscript review: Kingsmore SF
PI of the project (concept, design), subject recruitment, data analysis, manuscript editing and submission: Wallace MR
Data source and availabilityFull exome data from the four affected individuals will not be made available in a database, but we will share relevant data with researchers who have queries about genotypes at specific SNPs/genes, for modifier studies.
Financial support and sponsorshipFunding for this work was contributed by the American Heart Association (Southeast Affiliate) (MRW), the Hayward Foundation (MRW), University of Florida McNair Scholars Program (AS), and the Rady Children’s Institute for Genomic Medicine (SFK).
Conflicts of interestThe authors declare no conflicts of interest.
Patient consentHuman subject samples were obtained under IRB-approved consent.
Ethics approvalUniversity of Florida IRB approval was obtained (protocols 453-1992 and 2017-00320).
Copyright© The Author(s) 2018.
REFERENCES
1. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart hailure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 2006;113:1807-16.
3. Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagnostics 2013;15:158-67.
4. Konta L, Franklin RC, Kaski JP. Nomenclature and systems of classification for cardiomyopathy in children. Cardiol Young 2015;25:31-42.
5. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O' Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology task force on the definition and classification of cardiomyopathies. Circulation 1996;93:841-2.
6. Stone JR. Updates to the WHO classification of cardiomyopathies. Acad Forensic Pathol 2011;1:202-5.
7. Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res 2015;105:397-408.
8. Kostareva A, Kiselev A, Gudkova A, Frishman G, Ruepp A, Frishman D, Ruepp A, Frishman D, Smolina N, Tarnovskaya S, Nilsson D, Zlotina A, Khodyuchenko T, Versninina T, Pervunina T, Klyushina A, Kozlenok A, Sjoberg G, Golovjova I, Sejersen T, Shlyakhto E. Genetic spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS One 2016;11:e0163362.
9. Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol 2016;68:2871-86.
10. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Müller S, Kayvanpour E, Vogel B, Sedaghat-Hamedani F, Lim WK, Zhao X, Fradkin D, Köhler D, Fischer S, Franke J, Marquart S, Barb I, Li DT, Amr A, Ehlermann P, Mereles D, Weis T, Hassel S, Kremer A, King V, Wirsz E, Isnard R, Komajda M, Serio A, Grasso M, Syrris P, Wicks E, Plagnol V, Lopes L, Gadgaard T, Eiskjær H, Jørgensen M, Garcia-Giustiniani D, Ortiz-Genga M, Crespo-Leiro MG, Deprez RH, Christiaans I, van Rijsingen IA, Wilde AA, Waldenstrom A, Bolognesi M, Bellazzi R, Mörner S, Bermejo JL, Monserrat L, Villard E, Mogensen J, Pinto YM, Charron P, Elliott P, Arbustini E, Katus HA, Meder B. Atlas of the clinical genetics of dilated cardiomyopathy. Eur Heart J 2015;36:1123-35.
11. Gallego-Delgado M, Delgado JF, Brossa-Loidi V, Palomo J, Marzoa-Rivas R, Perez-Villa F, Salazar-Mendiguchia J, Ruiz-Cano MJ, Gonzalez-Lopez E, Padron-Barthe L, Bornstein B, Alonso-Pulpon L, Garcia-Pavia P. Idiopathic restrictive cardiomyopathy is primarily a genetic disease. J Am Coll Cardiol 2016;67:3021-3.
12. Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, Helio T, Keren A, McKenna WJ, Monserrat L, Pankuweit S, Perrot A, Rapezzi C, Ristic A, Seggewiss H, van Leangen I, Tavazzi L. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010;31:2715-26.
13. Ploski R, Rydzanicz M, Ksiazczyk TM, Franaszczyk M, Pollak A, Kosinska J, Michalak E, Stawinski P, Siolkowska L, Bilinska ZT, Werner B. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am J Med Genet A 2016;170:3241-8.
14. Yu HC, Coughlin CR, Geiger EA, Salvador BJ, Elias ER, Cavanaugh JL, Chatfield KC, Miyamoto SD, Shaikh TH. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Cold Spring Harb Mol Case Stud 2016;2:a000844.
15. Teo LY, Moran RT, Tang WH. Evolving approaches to genetic evaluation of specific cardiomyopathies. Curr Heart Failure Rep 2015;12:339-49.
17. Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W, Gibson WT, McKinnon ML, McGillivray B, Alvarez N, Giuffre M, Schwartzentruber J, FORGE Canada Consortium, Gerull B. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum Mutat 2016;37:269-79.
18. Wu W, Lu CX, Wang YN, Liu F, Chen W, Liu YT, Han YC, Cao J, Zhang SY, Zhang X. Novel phenotype-genotype correlations of restrictive cardiomyopathy with myosin-binding protein C (MYBPC3) gene mutations tested by next-generation sequencing. J Am Heart Assoc 2015;4:001879.
19. Zhang J, Kumar A, Kaplan L, Fricker FJ, Wallace MR. Genetic linkage of a novel autosomal dominant restrictive cardiomyopathy locus. J Med Genet 2005;42:663-5.
20. Zhang J, Kumar A, Stalker H, Virdi G, Ferrans V, Horiba K, Fricker FJ, Wallace M. Clinical and molecular studies of a large family with desmin-associated restrictive cardiomyopathy. Clin Genet 2001;59:248-56.
21. van der Ven PF, Obermann WM, Lemke B, Gautel M, Weber K, Fürst DO. Characterization of muscle filamin isoforms suggests a possible role of gamma-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil Cytoskeleton 2000;45:149-62.
22. Sethi R, Ylänne J. Small-angle X-ray scattering reveals compact domain-domain interactions in the n-terminal region of filamin C. PLoS One 2014;9:e107457.
23. Molt S, Bührdel JB, Yakovlev S, Schein P, Orfanos Z, Kirfel G, Winter L, Wiche G, van der Van PF, Rottbauer W, Just S, Belkin AM, Fürst DO. Aciculin interacts with filamin C and Xin and is essential for myofibril assembly, remodeling and maintenance. J Cell Sci 2014;127:3578-92.
24. Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padrón-Barthe L, Duro-Aguado I, Jiménez-Jáimez J, Hidalgo-Olivares VM, García-Campo E, Lanzillo C, Suárez-Mier MP, Yonath H, Marcos-Alonso S, Ochoa JP, Santomé JL, García-Giustiniani D, Rodríguez-Garrido JL, Domínguez F, Merlo M, Palomino J, Pe-a ML, Trujillo JP, Martín-Vila A, Stolfo D, Molina P, Lara-Pezzi E, Calvo-Iglesias FE, Nof E, Calò L, Barriales-Villa R, Gimeno-Blanes JR, Arad M, García-Pavía P, Monserrat L. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol 2016;68:2440-51.
25. Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, Reguero JR, Alvarez V, Moris C, Leon D, Martin M, Puente XS, López-Otín C. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun 2014;5:5326.
26. Leber Y, Ruparelia AA, Kirfel G, van der Ven PFM, Hoffmann B, Merkel R, Bryson-Richardson RJ, Fürst DO. Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. Hum Mol Genet 2016;25:2776-88.
27. Azzimato V, Genneback N, Tabish AM, Buyandelger B, Knoll R. Desmin, desminopathy, and the complexity of genetics. J Mol Cell Cardiol 2016;92:93-5.
28. Tucker NR, McLellan MA, Hu D, Ye J, Parsons VA, Mills RW, Clauss S, Dolmatova E, Shea MA, Milan DJ, Scott NS, Lindsay M, Lubitz SA, Domian IJ, Stone JR, Llin H, Ellinore PT. Novel mutation in FLNC (filamin C) causes familial restrictive cardiomyopathy. Circ Cardiovasc Genet 2017;10:e001780.
29. Gomez J, Lorca R, Reguero JR, Moris C, Martin M, Tranche S, Alonso B, Iglesias S, Alvarez V, Diaz-Molina B, Avanzas P, Coto E. Screening of the filamin C gene in a large cohort of hypertrophic cardiomyopathy patients. Circ Cardiovasc Genet 2017;10:e001584.
30. Avila-Smirnow D, Gueneau L, Batonnet-Pichon S, Delort F, Bécane H, Claeys, K., Beuvin M, Goudeau B, Jais JP, Nelson I, Richard P, Ben Yaou R, Romero NB, Wahbi K, Mathis S, Voit T, Furst D, van der Ven P, Gil R, Vicart P, Fardeau M, Bonne G, Behin A. Cardiac arrhythmia and late-onset muscle weakness caused by a myofibrillar myopathy with unusual histopathological features due to a novel missense mutation in FLNC. Neurolog Rev 2016;172:594-606.
31. Bang ML. Animal models of congenital cardiomyopathies associated with mutations in Z-line proteins. J Cell Physiol 2016;232:1-15.
32. Maerkens A, Kley RA, Olivé M, Theis V, van der Ven PF, Reimann J, Milting H, Schreiner A, Uszkoreit J, Eisenacher M, Barkovits K, Güttsches AK, Tonillo J, Kuhlmann K, Meyer HE, Schröder R, Tegenthoff M, Fürst DO, Müller T, Goldfarb LG, Vorgerd M, Marcus K. Differential proteomic analysis of abnormal intramyoplasmic aggregates in desminopathy. J Proteomics 2013;90:14-27.
33. Ton VK, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol 2015;8:39-44.
34. van den Bogaart FJ, Claeys KG, Kley R, Kusters B, Schrading S, Kamsteeg EJ, Voermans NC. Widening the spectrum of filamin-C myopathy: predominantly proximal myopathy due to the p.A193T mutation in the actin-binding domain of FLNC. Neuromusc Disorders 2017;27:73-7.
35. Ruparelia AA, Oorschot V, Ramm G, Bryson-Richardson RJ. FLNC myofibrillar myopathy results from impaired autophagy and protein insufficiency. Hum Mol Genet 2016;25:2131-42.
36. Jo BS, Koh IU, Bae JB, Yu HY, Jeon ES, Lee HY, Kim JJ, Choi M, Choi SS. Data of methylome and transcriptome derived from human dilated cardiomyopathy. Data Brief 2016;9:382-7.
37. Esslinger U, Garnier S, Korniat A, Proust C, Kararigas G, Müller-Nurasyid M, Empana JP, Morley MP, Perret C, Stark K, Bick AG, Prasad SK, Kriebel J, Li J, Tiret L, Strauch K, O'Regan DP, Marguiles KB, Seidman JG, Boutouyrie P, Lacolley P, Jouven X, Hengstenberg C, Komajda M, Hakonarson H, Isnard R, Arbustini E, Grallert H, Cook SA, Seidman CE, Regitz-Zagrosek V, Cappola TP, Charron P, Cambien F, Villard E. Exome-wide association study reveals novel susceptibility genes to sporadic dilated cardiomyopathy. PLoS One 2017;12:e0172995.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sanoja AJ, Li H, Fricker FJ, Kingsmore SF, Wallace MR. Exome sequencing identifies FLNC and ADD3 variants in a family with cardiomyopathy. J Transl Genet Genom 2018;2:6. http://dx.doi.org/10.20517/jtgg.2017.13
AMA Style
Sanoja AJ, Li H, Fricker FJ, Kingsmore SF, Wallace MR. Exome sequencing identifies FLNC and ADD3 variants in a family with cardiomyopathy. Journal of Translational Genetics and Genomics. 2018; 2: 6. http://dx.doi.org/10.20517/jtgg.2017.13
Chicago/Turabian Style
Sanoja, Alejandro J., Hua Li, F. Jay Fricker, Stephen F. Kingsmore, Margaret R. Wallace. 2018. "Exome sequencing identifies FLNC and ADD3 variants in a family with cardiomyopathy" Journal of Translational Genetics and Genomics. 2: 6. http://dx.doi.org/10.20517/jtgg.2017.13
ACS Style
Sanoja, AJ.; Li H.; Fricker FJ.; Kingsmore SF.; Wallace MR. Exome sequencing identifies FLNC and ADD3 variants in a family with cardiomyopathy. J. Transl. Genet. Genom. 2018, 2, 6. http://dx.doi.org/10.20517/jtgg.2017.13
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 14 clicks
Cite This Article 14 clicks

 Like This Article 26
likes
Like This Article 26
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.