
Genomics of cutaneous melanoma: focus on next-generation sequencing approaches and bioinformatics


Abstract
Cutaneous melanoma is caused by the uncontrolled growth of epidermal melanocytes. Melanoma continues to be a rare form of skin cancer but causes the majority of skin cancer related deaths. For many years the scientific community has focused on the investigation of the pathogenesis leading to melanoma, with the aim of better understanding its complexity and the potential advancement of therapeutic strategies. In this paper, the genomic features characterising the development of cutaneous melanoma are reviewed. Next-generation sequencing technologies and bioinformatics tools are currently state-of-the-art approaches in basic, applied and clinical cancer research. In this review, most of the available tools for revealing the mutational landscape are outlined.
Keywords
Introduction
Melanoma is a malignant tumour originating from melanocytes, the cells specialised to produce the melanin pigment. Melanocytes derive from the neural crest, a transient embryonic structure, consisting of highly migratory pluripotent cells, which give rise to a number of different cell types[1]. During development, melanocyte progenitors migrate, differentiate and colonize the skin-epidermis and hair follicles, the uvea of the eye and mucous membranes throughout the body. Accordingly, melanoma can arise at all these sites, leading to phenotypically, histologically, clinically and genetically diverse types of disease. In Caucasian populations, the most common type of melanoma is cutaneous melanoma (CM), originating from the epidermal melanocytes of non-glabrous skin. Among skin cancers, melanoma is the most aggressive, and although it accounts for less than 5% of skin cancer incidence, it is responsible for the majority of related deaths[2]. In this review we will focus on CM, as there are many differences in the genetic background implicated in different types of melanoma, such as mucosal or uveal melanoma. A distinct melanoma subtype, often referred to as a subtype of CM, is acral melanoma, occurring on glabrous (nonhair-bearing) acral skin of palms, soles and nail beds, which will not be further discussed in this review.
During the last decades, a continuous increase of CM frequency rates has been observed in Caucasian populations worldwide, making CM the cancer with the most rapidly increasing occurrence. CM incidence varies significantly between populations from different geographic regions, with Australia and New Zealand presenting the highest incidence rates worldwide. In Europe, rates are lower, but still have shown a three-fold to five-fold increase during this time period[3]. CM occurrence differs substantially between European countries, with Switzerland showing the highest rate and Greece belonging to the group of low-incidence countries[4,5].
CM development is a complex multi-factorial process, arising through multiple etiologic pathways and involving the interplay of genetic and environmental risk factors. Among them, the most well-established risk factors are exposure to ultraviolet (UV) radiation, family history, and phenotypic traits carrying a strong genetic component - including hair and eye colour, and the number of common and atypical melanocytic nevi[2].
In this review, we will summarize the progress towards the genomic characterization of CM, recent advances achieved through the exploitation of next-generation sequencing (NGS) technologies, as well as the bioinformatics tools developed for the analysis of sequencing data.
Germ-line susceptibility
Regarding the genetic background predisposing to melanoma, several susceptibility loci acting as high, moderate or low penetration genes, have been identified. Cyclin-dependent kinase inhibitor 2A (CDKN2A), the first familial melanoma gene identified[6,7], is found mutated in approximately 40% of melanoma high-density families. CDKN2A encodes for two distinct proteins, p16INK4A (p16) and p14ARF (p14), both involved in the regulation of the cell cycle[8]. The p16 and p14 mRNAs are transcribed from alternative first exons, so the related proteins have no similarity in their amino acid sequence, since they are translated in alternative reading frames. Mutations in p16 are predominantly loss of-function missense mutations, distributed throughout the protein, while in p14 inactivating mutations like whole gene deletions, insertions or splice-site mutations are mainly observed. Germ-line mutations in CDK4 are much less frequent and were initially identified by screening for p16 interacting partners. A mutational hotspot in codon 24, leading to an arginine substitution, abrogates the capacity of p16 to inactivate the kinase, thus promoting the G1-S phase transition. Other mutations have been identified in genes of more moderate penetrance, including BAP1, TERT, POT1, ACD, TERF2IP and MITF[9]. Genome-wide association studies have also revealed numerous recurring single nucleotide polymorphisms (SNPs) associated with melanoma risk[10-13].
Towards the genomic characterisation of melanoma
Identifying somatic mutations in the genome of melanoma is of great importance in order to understand the molecular basis of the disease’s genesis and progression. A number of oncogenes and tumour suppressor genes have been found to carry causative mutations. The first oncogene identified in melanoma was NRAS[14], which is also found mutated in other cancers. In 2002 the BRAF V600E somatic mutation was identified[15] and is the most frequent mutation found in CM patients. Since then, the advances in sequencing technology have enabled the application of massively parallel sequencing, thus dramatically changing our understanding of the somatic mutation landscape of melanoma. The first catalogue of somatic mutations of a cancer genome, at the whole-genome level concerned a melanoma cell line[16], indicated the presence of a great number of mutations per Mb and suggested a mutational signature related to UV exposure. Whole-exome sequencing studies exploiting clinical samples demonstrated that NF1, ARID2, PPP6C, RAC1, SNX31, TACC1, and STK19 are genes significantly mutated in melanoma[17,18]. The Cancer Genome Atlas Skin Cutaneous Melanoma (SKCM-TCGA) project confirmed, through exome sequencing, previously reported melanoma oncogenes and tumour suppressors (BRAF, NRAS, CDKN2A, TP53, and PTEN) and identified several additional significantly mutated melanoma genes, namely, MAP2K1, IDH1, RB1, and DDX3X[19]. The study proposed the classification of CM into four major genomic subtypes, related to the presence of specific mutations in established driver genes. In particular, the proposed genetic subtypes are the BRAF mutant, RAS mutant, NF1 mutant, and the triple wild-type. Low-frequency mutations were identified in the triple wild-type subtype in KIT, CTNNB1, GNA11, and GNAQ. More recently, the first large-scale study exploiting whole-genome sequencing supported the involvement of the non-coding genome in melanoma pathogenesis and revealed diverse carcinogenic processes across the different melanoma subtypes. Figure 1 summarizes the research on melanoma during the last decades, pinpointing key milestones in understanding its complexity.
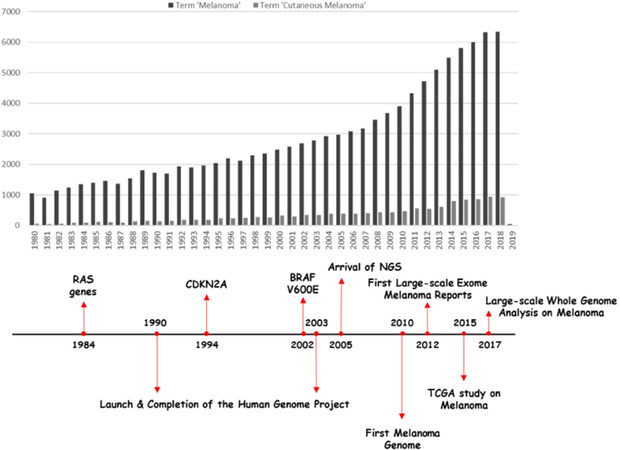
Figure 1. The number of publications per year on Pubmed (until 4th December 2018) using terms “melanoma” and “cutaneous melanoma” (upper), major landmarks concerning the study of melanoma (lower). NGS: next-generation sequencing
Mutation burden and specific signatures in melanoma
Sequencing of different cancers has revealed that the melanoma genome shows a substantial prevalence of somatic mutations[16,20,21]. Particularly, in CM an increased abundance of cytidine to thymidine (C > T) transitions is observed. This specific alteration is considered characteristic of a UV-light-induced mutational signature. A recent study, exploiting whole-genome sequencing of cutaneous, acral and mucosal melanomas, revealed distinct mutation profiles among these melanoma subtypes. The number of base substitutions and short insertions and/or deletions in CM was generally much higher than of those observed in acral and mucosal melanomas. In addition, the UV-related C > T transition was not observed in the latter melanoma subtypes. In contrast, somatic structural rearrangements were more frequent in acral and mucosal subtype[22]. These data suggest that different etiologic pathways are involved in the manifestation of diverse melanoma subtypes.
Next-generation sequencing approaches and bioinformatics
The significant progress towards the characterisation of the somatic mutational landscape of melanoma, can be mainly attributed to the rapid evolution of sequencing technologies during the last fifteen years. Nowadays, NGS has become the state-of-the-art tool in cancer research and is the most common and advanced technology for de novo somatic mutation detection. NGS technologies are in continuous development and improvement, both at the level of the applied protocols for library preparation and sequencing chemistry, but also at the bioinformatics level. A large number of bioinformatics tools have been developed for general pre-processing and basic analysis of NGS (WES/WGS) data with the aim of revealing altered variants for the cases under investigation. In this part of our review, we will focus only on tools developed for somatic mutation calling, bypassing those needed to reach this step of the analysis. Furthermore, we will discuss most of the available tools for driver-mutation identification, including the approaches that are used to achieve this step. Discriminating driver from passenger mutation remains a challenge from the experimental as well as the bioinformatics points of view[17,23-26]. In the case of melanoma, which is one of the cancers with the highest mutation burdens and heterogeneity, this problem is even more difficult to address, due to the confounding impact of melanoma’s high mutation rate. The aim of this review is not to perform a comparison of the tools since more detailed evaluations are available[26-28].
The basic approach for somatic variance identification is to compare paired samples, i.e., analyse matched tumour-normal samples collected from the same patient. Most callers are structured after this notion and use different approaches to extract the desired list of variants, meeting certain criteria. Among the strategies utilized are heuristic approaches combined with statistical tests, analysis and evaluation of a joint genotype likelihood, allele frequency or haplotype-based analyses, or exploitation of machine learning methods for variant classification. Apart from these, there are specialized tools that offer single-sample somatic mutation calling (lack of normal samples), through association with databases like COSMIC[29,30] and application of machine learning and statistical algorithms. Table 1 lists most somatic mutation callers based on their aforementioned strategic approaches.
Somatic variance calling tools
| Analysis tactic | Variant callers |
|---|---|
| Heuristic approaches | qSNP[31], RADIA[32], Shimmer[33], SOAPsnv[34], VarDict[35], VarScan2[36] |
| Joint genotype analysis | CaVEMan[37], FaSD-somatic[38], JointSNVMix2[39], SAMtools[40], Seurat*[41], SNVSniffer[42], SomaticSniper[43], Virmid[44] |
| Allele frequency | deepSNV[45], EBCall[46], LoFreq[47], LoLoPicker[48], MuTect[49], Strelka[50] |
| Haplotype analysis | FreeBayes[51], HapMuC[52], LocHap[53], MuTect2[49], Platypus[54] |
| Machine learning | BAYSIC[55], MutationSeq[56], SNooPer[57], SomaticSeq[58] |
| Single-sample analysis | GATKcan[59], ISOWN[60], OutLyzer[61], Pisces[62], SiNVICT[63], SomVarIUS[64] |
| Structural or copy number variation calling | APOLOH[65], BIC-Seq[66], BreakDancer[67], Break-Pointer[68], CNVkit[69], CoNIFER[70], Delly[71], HYDRA[72], GASV[73], GASVPro[74], Meerkat[65], PeSV-Fisher[75], VariationHunter-CommonLaw[76] |
| RNA-seq variant calling | eSNVdetect[77], SNPiR[78], VarDict[35], VarScan2[36] |
As a latter step, after obtaining a list of somatic mutations, it is important to distinguish the driver mutations which actively contribute to carcinogenesis[79]. This can be accomplished through mutation frequency analysis, functional impact investigation or machine learning algorithms based on known sets of driver/passenger genes. Another approach followed is enrichment analysis on known pathways or networks. Table 2 summarizes several tools which focus on driver mutation identification, classified by the strategic approach used. It is important to mention that distinction of driver/passenger genes faces many challenges mostly due to lack of annotation, additive effects of passenger mutations or a possible change in roles during cancer progression and the development of tumour heterogeneity[28]. In a recent publication, our group presented a methodology combining functional impact analysis with pathway enrichment, to deal with a limited dataset, in order to distinguish important genes and possible drivers exploiting exome sequencing data from melanoma patients in Greece[80].
Driver mutation calling tools
| Analysis tactic | Driver callers |
|---|---|
| Functional impact | CanPredict[81], Condel[82], FATHMM[83], GERP++[84], GOSS[85], MutationAssessor[86,87], MutationTaster[88], Oncodrive-fm[89], PMUT[90], PolyPhen-2[91], PROVEAN[92], SIFT[93], SNPs3D[94], TransFIC[95] |
| Mutation frequency | DrGaP[96], MuSiC[97], MutSig/MutSigCV[25], Youn and Simon[98] |
| Machine learning | CHASM[99,100], DMI[101] |
| Structural or copy number focus | ADMIRE[102], CMDS[103], GISTIC2[104], JISTIC[105] |
| Positional/structural clustering | iPAC[106], NMC[107] |
| Pathway/network analysis | BioInfoMiner[108], Dendrix[109], GSEA[110], HotNet[109], MEMo[111], Multi-Dendrix[112], NetBox[113], PathScan[114], Patient-oriented gene sets[115], RME[116] |
Genes bearing causative somatic mutations in melanoma
One of the most well-established pathways commonly affected in melanoma is the mitogen activating protein kinase (MAPK) signaling cascade, governing cell growth and survival. BRAF, NRAS and NF1 are the most frequently mutated genes of this pathway. Other pathways found significantly altered in CM include the phosphoinositide 3-kinase (PI3K) pathway, tumour protein 53 (TP53) signaling, cell cycle regulation and the telomere length maintenance pathway. In the next section, the most significant genes involved in such key processes, harboring driver mutations, are summarized.
BRAF
The BRAF gene encodes a serine/threonine protein kinase, belonging to the RAF family. This protein acts as a downstream effector of RAS-signalling in the MAPK cascade, affecting cell proliferation and survival. Mutations in this gene have been identified in various cancers. According to the COSMIC database 44% of melanomas arising from skin tissue have mutations in BRAF. In non-acral CM, the BRAF mutation of the kinase-activation domain at amino acid position 600, is the most common somatic mutation. Interestingly, BRAF V600E mutation results from a T→A transversion and not a C>T substitution, which is characteristic of UV light induced mutagenesis. Nevertheless, epidemiological and genomic evidence implies that UV radiation contributes to the formation of BRAF V600E. Soon after the characterization of BRAF V600E mutation in melanomas, it became apparent that its distribution greatly differs among different melanoma subtypes[117]. In particular, BRAF V600E mutations are more common in younger CM patients, whose melanomas arise on intermittently sun-exposed skin, on anatomical sites such as the trunk and proximal extremities. In contrast, melanomas arising on chronically sun-damaged skin, usually on anatomical sites like head, neck and the distal extremities of older individuals, have infrequent BRAF mutations, with BRAF V600K being more frequent than BRAF V600E[118]. Acral melanomas bear BRAF mutations much less frequently. Targeting the BRAF-V600E mutant protein with specific inhibitors exposed new therapeutic aspects for the management of such an aggressive disease. The oncogenic activation of BRAF mutations is considered a necessary but not sufficient condition to transform melanocytes to melanoma cells, a suggestion which is also supported by the frequent occurrence of such mutations in benign nevi[119].
RAS
RAS proteins are small GTPases functioning as GDP-GTP-regulated binary switches that control many fundamental cellular processes. RAS proteins connect a great variety of upstream signals from activated membrane receptors to downstream pathways controlling the cell cycle, growth, apoptosis, and senescence[119]. The HRAS, KRAS, and NRAS oncogenes were the first human oncogenes to be discovered[120]. In the case of CM, NRAS mutations are found in 17% of the cases, according to COSMIC database[29]. NRAS hot-spot mutations are mutually exclusive of BRAF hot-spot mutations. HRAS and KRAS mutations are much less frequent in CM. Regarding NRAS, the most common mutations cause a change of the amino acid at position 61, lying at the GTP-binding domain. These substitutions disrupt the GTPase activity of the protein, locking it in its active conformation[121].
NF1
NF1 is a tumour suppressor gene encoding for a direct negative regulator of RAS signaling[122]. In particular, NF1 is a GTPase-activating protein known to downregulate RAS activity by stimulating the hydrolysis of GTP and returning the protein to its inactive form. A significant enrichment of NF1 mutations was found in BRAF and NRAS wild-type melanomas[17,18]. In the TCGA study NF1 was found as the third most frequently observed significantly mutated gene of the MAPK pathway[123]. Mutations in the NF1 gene are loss of function mutations, mainly nonsense point mutations[124], which can be considered as an alternative way to activate the MAPK signaling pathway.
TERT
The TERT gene encodes for the telomerase reverse transcriptase, the catalytic subunit of the telomerase ribonucleoprotein, essential for the maintenance of telomeres and chromosomal stability. Recurrent somatic mutations in the TERT promoter have been characterised in CM, with high frequency in sporadic melanoma. Specifically, the two hot-spot mutations, located at -124 and -146bp relative to the transcriptional start site, are C>T transitions, consistent with a UV signature mutational profile[125,126]. In a recent study exploiting whole genome sequencing, 86% of CM cases were found mutated at one or more out of four positions upstream of the transcriptional start site[22]. All these mutations are mutually exclusive and create new binding sites for the E26 transformation-specific family transcription factor GA-binding protein. Recent evidence suggests that TERT promoter mutations result in TERT over expression[127,128]. They are established after MAPK-pathway activating mutations, but still during the early stage of melanoma progression[129].
CDKN2A
CDKN2A is a well characterised tumour-suppressor gene, found to harbour somatic alterations in a wide variety of different tumour types[128]. Regarding CM, in addition to its association with familial melanoma, somatic alterations resulting in CDKN2A inactivation are also frequently observed in sporadic melanoma. The most frequent alteration is the deletion of the CDKN2A gene, reported in 41% of CM[129]. CDKN2A expression is additionally regulated at the epigenetic level, mainly by methylation of its promoter and subsequent gene silencing. The two proteins encoded by CDKN2A, p16 and p14, have distinct roles in the regulation of the cell cycle. p16 modulates G1 to S phase transition by inhibiting the kinase activity of cyclin dependent kinases 4 and 6 (CDK4 and CDK6), while p14 acts through TP53 stabilisation. Biallelic loss of CDKN2A and subsequent disruption of the G1/S checkpoint, is believed to be a crucial step in melanoma progression towards transition to the invasive phenotype[127].
TP53
TP53 is a well-known tumour suppressor gene, involved in the transcriptional regulation of several target genes. TP53 is mutated in 27 different types of cancer[130]. Regarding melanoma, 15% of cases harbour mutations in TP53[29]. Based on mutational studies, comparing primary melanomas and metastases, TP53 was found to be more frequently mutated in melanoma metastases, indicating that TP53 mutations may arise later during melanoma progression[118].
PTEN
PTEN is a tumour-suppressor gene, coding for the phosphatidyl-inositol-3,4,5-triphosphate 3-phosphatase. PTEN phosphatase is a fundamental regulator of the PI3K/AKT pathway, exerting its inhibitory effects on AKT signaling, by dephosphorylating PIP3. PIP3 acts as a second messenger, triggering a number of signaling cascades- among them AKT- which play a key role in processes like cell survival and proliferation, apoptosis, and cellular metabolism[131]. Somatic mutations in PTEN, primarily deletions but also loss-of-function SNVs - 18 % and 8% respectively in CM[129], result in PTEN inactivation and promote cell survival through sustained activation of the PI3K signaling pathway[132].
MITF
Microphthalmia-associated transcription factor (MITF) is a basic helix-loop-helix/leucine zipper transcription factor required for melanocyte development. MITF is essential for establishing the melanocytic lineage during differentiation of neural crest cells[133]. Transcriptional targets of MITF, include genes encoding for components of melanosomes, enzymes of the melanin synthesis pathway, as well as genes involved in cell cycle regulation and cell survival. Somatic amplification of MITF has been identified in melanomas, but MITF activity is mainly altered by its upstream activators and suppressors acting on the transcriptional, post-transcriptional and post-translational levels.
Other genes in melanomagenesis
Other genes causatively implicated in melanomagenesis and progression include KIT, RAC1 and ARID2. KIT encodes for a tyrosine kinase, which is the receptor of the Stem Cell Factor. Upon ligand binding, multiple signalling pathways affecting cell growth, proliferation, survival, and migration are activated. In CM, mutations in KIT occur most commonly in melanomas originating from chronically sun damaged skin and in the acral subtype[118]. The RAC1 gene encodes for a GTPase of the Ras superfamily with important roles in cell motility. A hot spot mutation at P29S, is the result of a C>T transition, consistent with the molecular signature associated with UV damage[18]. The ARID2 gene encodes for a subunit of the switch/sucrose non-fermentable (SWI/SNF) chromatin remodelling complex, a multiprotein complex that alters chromatin structure to regulate gene expression[134]. Recent evidence suggests that components of the SWI/SNF complex, function as tumour suppressors in several types of cancer. In the case of CM, loss-of-function mutations in the ARID2 gene are the most frequent among SWI/SNF enzymes.
Conclusion
In this review, we present the main genetic features contributing to the development of CM. Marked advances in dealing with this complex disease have been achieved over the last years, due to the diligent efforts of researchers to shed light on the biological mechanisms involved in melanoma manifestation, assisted by the advent of NGS technologies. Elucidating the mechanisms underlying melanoma biology and progression can enable the development of targeted and immune-related therapeutic approaches. Still, melanoma remains one of the most lethal types of cancer. Additional understanding of the resistance to targeted therapies is crucial, and ought to remain a central aspect of cancer research. The intervention schemes based on combination approaches are the most promising therapeutic ways, in the context of personalised treatment.
Declarations
AcknowledgmentsThe authors would like to thank Dr. S. Kyrtopoulos and G. Sotiroudis, MSc, for proofreading this paper.
Authors’ contributionsDesign: Papadodima O, Maglogiannis I, Chatziioannou A
Literature research: Papadodima O, Kontogianni G, Pyroti G, Chatziioannou A
Manuscript writing, editing, revision: Papadodima O, Kontogianni G, Pyroti G, Maglogiannis I, Chatziioannou A
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by “TRANSITION: TRANSlating the complexIty of melanoma diagnosis into raTional therapeutic stratificatiON” (MIS 5031295) which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
2. Nikolaou V, Stratigos A. Emerging trends in the epidemiology of melanoma. Br J Dermatol 2014;170:11-9.
4. ECIS- European Cancer Information System. Available from: https://ecis.jrc.ec.europa.eu [Last accessed on 26 Feb 2019].
5. Forsea AM, Del Marmol V, De Vries E, Bailey E, Geller A. Melanoma incidence and mortality in Europe: new estimates, persistent disparities. Br J Dermatol 2012;167:1124-30.
6. Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, et al. Germline p16 mutations in familial melanoma. Nature genetics 1994;8:15-21.
7. Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nature genetics 1994;8:23-6.
8. de Snoo FA, Hayward NK. Cutaneous melanoma susceptibility and progression genes. Cancer Lett 2005;230:153-86.
9. Aoude LG, Wadt KA, Pritchard AL, Hayward NK. Genetics of familial melanoma: 20 years after CDKN2A. Pigment Cell Melanoma Res 2015;28:148-60.
10. Athanasiadis EI, Antonopoulou K, Chatzinasiou F, Lill CM, Bourdakou MM, et al. A Web-based database of genetic association studies in cutaneous melanoma enhanced with network-driven data exploration tools. Database (Oxford) 2014;2014:bau101.
11. Antonopoulou K, Stefanaki I, Lill CM, Chatzinasiou F, Kypreou KP, et al. Updated field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma: the MelGene database. J Invest Dermatol 2015;135:1074-9.
12. Chatzinasiou F, Lill CM, Kypreou K, Stefanaki I, Nicolaou V, et al. Comprehensive field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma. J Natl Cancer Inst 2011;103:1227-35.
13. Law MH, Bishop DT, Lee JE, Brossard M, Martin NG, et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nature genetics 2015;47:987-95.
14. Padua RA, Barrass N, Currie GA. A novel transforming gene in a human malignant melanoma cell line. Nature 1984;311:671-3.
15. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54.
16. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463:191-6.
17. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251-63.
18. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nature genetics 2012;44:1006-14.
19. The Cancer Genome Atlas [Internet]. Available from: https://cancergenome.nih.gov/. [Last accessed on 24 Feb 2019].
20. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007;446:153-8.
21. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415-21.
22. Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017;545:175-80.
23. Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res 2012;40:e169.
24. Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, et al. IntOGen-mutations identifies cancer drivers across tumor types. Nature methods 2013;10:1081-2.
25. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214-8.
26. Raphael BJ, Dobson JR, Oesper L, Vandin F. Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome medicine 2014;6:5.
27. Xu C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput Struct Biotechnol J 2018;16:15-24.
28. Zhang J, Liu J, Sun J, Chen C, Foltz G, et al. Identifying driver mutations from sequencing data of heterogeneous tumors in the era of personalized genome sequencing. Brief Bioinformatics 2014;15:244-55.
29. Cosmic. COSMIC - Catalogue of Somatic Mutations in Cancer [Internet]. Available from: https://cancer.sanger.ac.uk/cosmic. [Last accessed on 24 Feb 2019].
30. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 2017;45:D777-83.
31. Kassahn KS, Holmes O, Nones K, Patch AM, Miller DK, et al. Somatic point mutation calling in low cellularity tumors. PLoS One 2013;8:e74380.
32. Radenbaugh AJ, Ma S, Ewing A, Stuart JM, Collisson EA, et al. RADIA: RNA and DNA integrated analysis for somatic mutation detection. PLoS One 2014;9:e111516.
33. Hansen NF, Gartner JJ, Mei L, Samuels Y, Mullikin JC. Shimmer: detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics 2013;29:1498-503.
34. SOAP : Short Oligonucleotide Analysis Package [Internet]. Available from: http://soap.genomics.org.cn/. [Last accessed on 24 Feb 2019].
35. Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 2016;44:e108.
36. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568-76.
37. Jones D, Raine KM, Davies H, Tarpey PS, Butler AP, et al. cgpCaVEManWrapper: Simple Execution of CaVEMan in Order to Detect Somatic Single Nucleotide Variants in NGS Data. Curr Protoc Bioinformatics 2016;56:15.10.1-15.10.18.
38. Wang W, Wang P, Xu F, Luo R, Wong MP, et al. FaSD-somatic: a fast and accurate somatic SNV detection algorithm for cancer genome sequencing data. Bioinformatics 2014;30:2498-500.
39. Roth A, Ding J, Morin R, Crisan A, Ha G, et al. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 2012;28:907-13.
40. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011;27:2987-93.
41. Christoforides A, Carpten JD, Weiss GJ, Demeure MJ, Von Hoff DD, et al. Identification of somatic mutations in cancer through Bayesian-based analysis of sequenced genome pairs. BMC Genomics 2013;14:302.
42. Liu Y, Loewer M, Aluru S, Schmidt B. SNVSniffer: an integrated caller for germline and somatic single-nucleotide and indel mutations. BMC Syst Biol 2016;10:47.
43. Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012;28:311-7.
44. Kim S, Jeong K, Bhutani K, Lee J, Patel A, et al. Virmid: accurate detection of somatic mutations with sample impurity inference. Genome Biol 2013;14:R90.
45. Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun 2012;3:811.
46. Shiraishi Y, Sato Y, Chiba K, Okuno Y, Nagata Y, et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res 2013;41:e89.
47. Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 2012;40:11189-201.
48. Carrot-Zhang J, Majewski J. LoLoPicker: detecting low allelic-fraction variants from low-quality cancer samples. Oncotarget 2017;8:37032-40.
49. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213-9.
50. Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811-7.
51. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. Quantitative Biology 2012.
52. Usuyama N, Shiraishi Y, Sato Y, Kume H, Homma Y, et al. HapMuC: somatic mutation calling using heterozygous germ line variants near candidate mutations. Bioinformatics 2014;30:3302-9.
53. Sengupta S, Gulukota K, Zhu Y, Ober C, Naughton K, et al. Ultra-fast local-haplotype variant calling using paired-end DNA-sequencing data reveals somatic mosaicism in tumor and normal blood samples. Nucleic Acids Res 2016;44:e25.
54. Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SRF, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 2014;46:912-8.
55. Cantarel BL, Weaver D, McNeill N, Zhang J, Mackey AJ, et al. BAYSIC: a Bayesian method for combining sets of genome variants with improved specificity and sensitivity. BMC Bioinformatics 2014;15:104.
56. Ding J, Bashashati A, Roth A, Oloumi A, Tse K, et al. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics 2012;28:167-75.
57. Spinella JF, Mehanna P, Vidal R, Saillour V, Cassart P, et al. SNooPer: a machine learning-based method for somatic variant identification from low-pass next-generation sequencing. BMC Genomics 2016;17:912.
58. Fang LT, Afshar PT, Chhibber A, Mohiyuddin M, Fan Y, et al. An ensemble approach to accurately detect somatic mutations using SomaticSeq. Genome Biol 2015;16:197.
59. Hsu YC, Hsiao YT, Kao TY, Chang JG, Shieh GS. Detection of somatic mutations in exome sequencing of tumor-only samples. Sci Rep 2017;7:15959.
60. Kalatskaya I, Trinh QM, Spears M, McPherson JD, Bartlett JMS, et al. ISOWN: accurate somatic mutation identification in the absence of normal tissue controls. Genome Med 2017;9:59.
61. Muller E, Goardon N, Brault B, Rousselin A, Paimparay G, et al. OutLyzer: software for extracting low-allele-frequency tumor mutations from sequencing background noise in clinical practice. Oncotarget 2016;7:79485-93.
62. Dunn T, Berry G, Emig-Agius D, Jiang Y, Lei S, et al. Pisces: an accurate and versatile variant caller for somatic and germline next-generation sequencing data. Bioinformatics 2018; doi: 10.1093/bioinformatics/bty849.
63. Kockan C, Hach F, Sarrafi I, Bell RH, McConeghy B, et al. SiNVICT: ultra-sensitive detection of single nucleotide variants and indels in circulating tumour DNA. Bioinformatics 2017;33:26-34.
64. Smith KS, Yadav VK, Pei S, Pollyea DA, Jordan CT, et al. SomVarIUS: somatic variant identification from unpaired tissue samples. Bioinformatics 2016;32:808-13.
65. Yang L, Luquette LJ, Gehlenborg N, Xi R, Haseley PS, et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 2013;153:919-29.
66. Xi R, Luquette J, Hadjipanayis A, Kim TM, Park PJ. BIC-seq: a fast algorithm for detection of copy number alterations based on high-throughput sequencing data. Genome Biol 2010;11:O10.
67. Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, et al. BreakDancer: an algorithm for high resolution mapping of genomic structural variation. Nat Methods 2009;6:677-81.
68. Drier Y, Lawrence MS, Carter SL, Stewart C, Gabriel SB, et al. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res 2013;23:228-35.
69. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS computational biology 2016;12:e1004873.
70. Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res 2012;22:1525-32.
71. Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28:i333-9.
72. Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, et al. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res 2013;23:762-76.
73. Sindi S, Helman E, Bashir A, Raphael BJ. A geometric approach for classification and comparison of structural variants. Bioinformatics 2009;25:i222-30.
74. Sindi SS, Önal S, Peng LC, Wu HT, Raphael BJ. An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol 2012;13:R22.
75. Escaramís G, Tornador C, Bassaganyas L, Rabionet R, Tubio JMC, et al. PeSV-Fisher: identification of somatic and non-somatic structural variants using next generation sequencing data. PLoS One 2013;8:e63377.
76. Hormozdiari F, Hajirasouliha I, McPherson A, Eichler EE, Sahinalp SC. Simultaneous structural variation discovery among multiple paired-end sequenced genomes. Genome Res 2011;21:2203-12.
77. Tang X, Baheti S, Shameer K, Thompson KJ, Wills Q, et al. The eSNV-detect: a computational system to identify expressed single nucleotide variants from transcriptome sequencing data. Nucleic Acids Res 2014;42:e172.
78. Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet 2013;93:641-51.
80. Kontogianni G, Piroti G, Maglogiannis I, Chatziioannou A, Papadodima O. Dissecting the mutational landscape of cutaneous melanoma: an omic analysis based on patients from Greece. Cancers 2018;10:96.
81. Kaminker JS, Zhang Y, Watanabe C, Zhang Z. CanPredict: a computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res 2007;35:W595-8.
82. González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 2011;88:440-9.
83. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Human mutation 2013;34:57-65.
84. Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 2010;6:e1001025.
85. Kaminker JS, Zhang Y, Waugh A, Haverty PM, Peters B, et al. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res 2007;67:465-73.
86. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011;39:e118.
87. Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol 2007;8:R232.
88. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361-2.
89. Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res 2012;40:e169.
90. Ferrer-Costa C, Gelpí JL, Zamakola L, Parraga I, de la Cruz X, et al. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 2005;21:3176-8.
91. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. A method and server for predicting damaging missense mutations. United States: Nat Methods; 2010. pp. 248-9.
92. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One 2012;7:e46688.
94. Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics 2006;7:166.
95. Gonzalez-Perez A, Deu-Pons J, Lopez-Bigas N. Improving the prediction of the functional impact of cancer mutations by baseline tolerance transformation. Genome Med 2012;4:89.
96. Hua X, Xu H, Yang Y, Zhu J, Liu P, et al. DrGaP: a powerful tool for identifying driver genes and pathways in cancer sequencing studies. Am J Hum Genet 2013;93:439-51.
97. Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res 2012;22:1589-98.
98. Youn A, Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics 2011;27:175-81.
99. Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res 2009;69:6660-7.
100. Wong WC, Kim D, Carter H, Diekhans M, Ryan MC, et al. CHASM and SNVBox: toolkit for detecting biologically important single nucleotide mutations in cancer. Bioinformatics 2011;27:2147-8.
101. Tan H, Bao J, Zhou X. A novel missense-mutation-related feature extraction scheme for “driver” mutation identification. Bioinformatics 2012;28:2948-55.
102. van Dyk E, Reinders MJT, Wessels LFA. A scale-space method for detecting recurrent DNA copy number changes with analytical false discovery rate control. Nucleic Acids Res 2013;41:e100.
103. Zhang Q, Ding L, Larson DE, Koboldt DC, McLellan MD, et al. CMDS: a population-based method for identifying recurrent DNA copy number aberrations in cancer from high-resolution data. Bioinformatics 2010;26:464-9.
104. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12:R41.
105. Sanchez-Garcia F, Akavia UD, Mozes E, Pe’er D. JISTIC: Identification of significant targets in cancer. BMC Bioinformatics 2010;11:189.
106. Ryslik GA, Cheng Y, Cheung KH, Modis Y, Zhao H. Utilizing protein structure to identify non-random somatic mutations. BMC Bioinformatics 2013;14:190.
107. Ye J, Pavlicek A, Lunney EA, Rejto PA, Teng CH. Statistical method on nonrandom clustering with application to somatic mutations in cancer. BMC Bioinformatics 2010;11:11.
108. Koutsandreas T, Binenbaum I, Pilalis E, Valavanis I, Papadodima O, et al. Analyzing and visualizing genomic complexity for the derivation of the emergent molecular networks. IJMSTR 2016;4:30-49.
109. Vandin F, Upfal E, Raphael BJ. Algorithms for detecting significantly mutated pathways in cancer. J Comput Biol 2011;18:507-22.
110. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545-50.
111. Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res 2012;22:398-406.
112. Leiserson MDM, Blokh D, Sharan R, Raphael BJ. Simultaneous identification of multiple driver pathways in cancer. PLoS Comput Biol 2013;9:e1003054.
113. Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PLoS One 2010;5:e8918.
114. Wendl MC, Wallis JW, Lin L, Kandoth C, Mardis ER, et al. PathScan: a tool for discerning mutational significance in groups of putative cancer genes. Bioinformatics 2011;27:1595-602.
115. Boca SM, Kinzler KW, Velculescu VE, Vogelstein B, Parmigiani G. Patient-oriented gene set analysis for cancer mutation data. Genome Biol 2010;11:R112.
116. Miller CA, Settle SH, Sulman EP, Aldape KD, Milosavljevic A. Discovering functional modules by identifying recurrent and mutually exclusive mutational patterns in tumors. BMC Med Genomics 2011;4:34.
117. Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst 2003;95:1878-90.
118. Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol 2014;9:239-71.
119. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell 2017;170:17-33.
121. Fedorenko IV, Gibney GT, Smalley KSM. NRAS mutant melanoma: biological behavior and future strategies for therapeutic management. Oncogene 2013;32:3009-18.
122. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 2010;10:842-57.
124. Cirenajwis H, Lauss M, Ekedahl H, Törngren T, Kvist A, et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol Oncol 2017;11:438-51.
125. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, et al. Highly recurrent TERT promoter mutations in human melanoma. Science 2013;339:957-9.
126. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013;339:959-61.
127. Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, et al. Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell 2018;34:45-55.e4.
128. Zhao R, Choi BY, Lee MH, Bode AM, Dong Z. Implications of genetic and epigenetic alterations of CDKN2A (p16(INK4a)) in cancer. EBioMedicine 2016;8:30-9.
129. Palmieri G, Colombino M, Casula M, Manca A, Mandalà M, et al. Molecular pathways in melanomagenesis: what we learned from next-generation sequencing approaches. Curr Oncol Rep 2018;20:86.
130. Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018;173:371-85.e18.
131. Ortega-Molina A, Serrano M. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab 2013;24:184-9.
132. Helgadottir H, Rocha Trocoli Drakensjö I, Girnita A. Personalized medicine in malignant melanoma: towards patient tailored treatment. Front Oncol 2018;8:202.
133. Mort RL, Jackson IJ, Patton EE. The melanocyte lineage in development and disease. Development 2015;142:620-32.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Papadodima O, Kontogianni G, Piroti G, Maglogiannis I, Chatziioannou A. Genomics of cutaneous melanoma: focus on next-generation sequencing approaches and bioinformatics. J Transl Genet Genom 2019;3:7. http://dx.doi.org/10.20517/jtgg.2018.33
AMA Style
Papadodima O, Kontogianni G, Piroti G, Maglogiannis I, Chatziioannou A. Genomics of cutaneous melanoma: focus on next-generation sequencing approaches and bioinformatics. Journal of Translational Genetics and Genomics. 2019; 3: 7. http://dx.doi.org/10.20517/jtgg.2018.33
Chicago/Turabian Style
Papadodima, Olga, Georgia Kontogianni, Georgia Piroti, Ilias Maglogiannis, Aristotelis Chatziioannou. 2019. "Genomics of cutaneous melanoma: focus on next-generation sequencing approaches and bioinformatics" Journal of Translational Genetics and Genomics. 3: 7. http://dx.doi.org/10.20517/jtgg.2018.33
ACS Style
Papadodima, O.; Kontogianni G.; Piroti G.; Maglogiannis I.; Chatziioannou A. Genomics of cutaneous melanoma: focus on next-generation sequencing approaches and bioinformatics. J. Transl. Genet. Genom. 2019, 3, 7. http://dx.doi.org/10.20517/jtgg.2018.33
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 25 clicks
Cite This Article 25 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.