
Intellectual disability, the long way from genes to biological mechanisms
 0
0Abstract
Approximately 2% of the world population is affected by intellectual disability (ID). Huge efforts in sequencing and analysis of individual human genomes have identified several genes and genetic/genomic variants associated with ID. Despite all this knowledge, the relationship between genes, pathophysiology and molecular mechanisms of ID remain highly complex. We summarize the genomic advances related to ID, provide examples on how to discern correlative versus causative roles in genetic variation, understand the physiological consequences of identified variants, and discuss future challenges.
Keywords
Introduction
Processes such as memory, attention, reasoning and executive function are collectively embedded in the concept of cognition. While cognitive abilities in humans are variable and inheritable[1], identification of the genetic determinants of human cognition has been limited. Candidate genes involved in the molecular underpinnings of cognition can be identified through studies on cognitive disorders. The impairment of cognitive function is a core clinical feature of neurodevelopmental disorders (NDDs), which comprise a group of developmental disorders leading to brain dysfunction. NDDs include global developmental delay, intellectual disability (ID), schizophrenia, autism spectrum disorder, attention-deficit/hyperactivity disorder, bipolar disorder, and epilepsy. Studies on NDDs have revealed that cognitive disorders are complex, usually polygenic[2], and phenotypically and genetically heterogeneous[3,4]. ID is characterized by significant limitations in both intellectual functioning and in adaptive behavior including conceptual, social, and practical adaptive skills[5].
ID originates during the developmental period and has an incidence of ~2% in the population[6-8]. Although ID can be caused by environmental factors such as maternal alcohol abuse during pregnancy, infections, birth complications and extreme malnutrition, genetic factors are now known to have an important role in its etiology, accounting for the majority of cases. ID is the most common reason for referral to genetic services and recent technological advances have allowed genetic diagnoses to be obtained for a substantial portion of affected individuals. The combination of novel technologies and increased biological understanding is rapidly increasing the diagnostic yield of genetic tests in ID. The introduction of chromosome array analysis (comparative genomic hybridization, CGH) has allowed the genome-wide detection of chromosomal aberrations, while exome sequencing (WES) and more recently whole genome sequencing (WGS) have enabled testing of all genes simultaneously in a single test. Currently, WGS is becoming the first-tier diagnostic test, which also allows for the detection of chromosomal aberrations[9]. These are impressive advancements that have important ramifications for both treatment and prognosis. A specific diagnosis also provides both psychological and social benefits for the family[10], including information about the risk of recurrence in future pregnancies and the options of prenatal diagnosis and pre-implantation genetic testing. As of December 2019, on the Online Mendelian Inheritance in Man website (OMIM, https://omim.org/), there are more than 1300 single genes associated with ID, highlighting the complexities of brain development and the consequent, extreme genetic heterogeneity of ID. These genes are all related to a variety of cellular functions and molecular processes. On top of the functional diversity of ID associated genes, there is a myriad of genetic variants within the same gene loci with different pathological consequences, ranging from benign (no identifiable phenotypic consequences) to clearly pathogenic (associated with extreme phenotypic outcomes). Identification of new genes and genetic variants related to ID and improved understanding of the biological functions associated with these mutations are now critical.
Genomic advances related to ID
During the late twentieth century, twin studies showed that ID has a strong heritable component[11]. However, only in the beginning of the new millennium, with the advent of Next Generation- or massive- Sequencing technologies, has determination of the underlying genetic cause of ID, as well as many other congenital diseases, become possible[12]. An accurate molecular diagnosis is essential for the optimization of clinical management and the institution of appropriate surveillance and prevention programs[13]. De novo mutations account for at least 30%, and possibly as much as 60%, of ID cases, with diagnostic efficiency in clinical practice around 25%-30%[14]. This low diagnostic yield begets the question of what the causes of ID are in the remaining 75% of patients. Genetic and phenotypic variability, and the non-specific nature of the phenotype makes accurate genetic diagnosis in the majority of children with ID a very challenging task. In cases where no obvious causes are found, the differential diagnosis can include hundreds of rare genetic disorders, leading to hundreds of potentially involved genes, with both single nucleotide (SNVs) and copy-number variants (CNVs) putatively contributing to disease development. In this context, different molecular techniques for diagnosis coexist with each having particular pros and cons[15-17].
Array based CGH was the first choice for diagnosing ID ten years ago, with a two-fold increase in diagnostic yield compared with karyotype analysis[15]. CGH allowed precise identification of CNVs as small as 20 kb long, including heterozygous deletions and duplications. However, a significant number of patients remained undiagnosed and consequently, physicians moved on to target sequencing of disease associated genes, or more recently WES. Target sequencing and WES allow identification of SNVs as well as small indels (2-20 bp) providing a diagnostic yield of 25% for children with ID[18]. The main difference between target and WES is the per sample cost, which tends to be lower for the target approach[19]. However, due to the aforementioned high phenotypic variability, sequencing only a limited number of genes can reduce the overall diagnostic yield.
The reduction in sequencing costs in the last decades has enabled WGS to be added to the diagnostic armamentarium. WGS has the potential to identify all forms of genetic variants, SNVs, indels, as well as CNVs. Recent studies demonstrated the advantages of WGS over both CGH and WES for the identification of novel mutations, with an overall diagnostic yield between 40%-60% for children with ID. The genetic heterogeneity of ID[17,20] makes WGS possibly the most cost-effective approach in terms of diagnostic yield and sequencing costs. However, it is important to note that WGS has larger costs related to data processing and storage, as well as analysis - which is much more challenging - compared to CGH or WES. As an example, while WES provides about 100,000 SNVs, WGS yields over 3 million variants per sample, of which only one (or a few) are likely to be relevant to the case. Moreover, WGS will require appropriate counseling, including appropriate management of any incidental finding.
Interpretation of genetic variants in ID patients
One of the main challenges in the molecular diagnosis of ID concerns the identification and, most importantly, assignment of any found variant as responsible for the observed phenotype. This task, which requires the annotation, interpretation and selection of variants for each case, is usually performed in a multidisciplinary context, with the involvement of bioinformaticians, molecular geneticists and the responsible physician, and is referred to as Clinical Genomics Interpretation. The complexity of the task is related to the chosen technique. While sequencing of gene panels - already focused on ID associated genes - delivers a few hundred variants, CGH results in thousands of CNVs, WES yields up to 100,000 variants, and finally for WGS, over a million.
The first step of variant filtering (i.e., reduction of the number of potential candidates) involves focusing on ID related genes. OMIM, an Online Catalog of Human Genes and Genetic Disorders, lists 1330 independent genes associated with the words “intellectual disability” - double the number of ID-related genes listed in 2015 - with a variety of functions and modes of inheritance[21]. The latest update on 4 December 2019 of the[22] SysID-database (https://sysid.cmbi.umcn.nl/) currently contains 1291 primary ID genes, and 1140 candidate ID genes. This huge number and functional diversity of ID-related genes contributes to the challenge of identifying new genes or genetic variants related to ID unequivocally.
Another important filtering criterion is related to genetic variation properties. Current state of the art techniques classify variants according to the American College of Medical Genetics criteria[23], which involves determination of several evidence criteria (or level), which then add up to a final score that determines whether the variant is (likely) benign, (likely) pathogenic or of uncertain significance (i.e., a variant of unknown significance or VoUS). Although there are more than 25 different criteria, they can arguably be grouped into those related to: (1) predicted molecular effect; (2) observed frequency in healthy individuals; (3) familial segregation; (4) genotype to phenotype relationships; and (5) previous reports. Ideally, the combination of genomic techniques and use of appropriate filtering criteria should result in the identification and report of a (likely) pathogenic variant. Yet, as will be explained below, this is particularly difficult when dealing with ID related variants. Databases such as IDGenetics, (http://www.ccgenomics.cn/IDGenetics/)[24], a genetic database for ID that provides integrated genetic, genomic and biological data, can facilitate the interpretation of ID related genetic variants.
To be classified as (likely) pathogenic, a variant would usually have a strong molecular effect (i.e., nonsense, frameshift, affect splicing and/or missense with a known molecular phenotype), display very low population frequency, verified to be de novo, display a known matching phenotype and have been previously reported in ID or NDD case. However, since most ID variants are de novo, they are also novel, and thus unlikely to have been reported and/or studied at the molecular level, particularly if they are missense SNVs. For example, one of the most commonly mutated genes in patients with ID, ARID1B, comprises only 1% of all ID cases. Moreover, since there are many associated genes and the phenotype in ID patients is highly variable and overlapping, it is extremely difficult to decide between variants with similar evidence criteria but located in different genes. For CNV, where genomic intervals deviate from the normal diploid state, the molecular effect is easier to gauge since the whole gene (or a significant part of it) is usually deleted or duplicated, conferring a gene dosage effect. CNVs are also more likely to be unique for the patient but there are some hot spot CNVs, mainly the ones related to syndromic ID, such as the 7q11.23 deletion that is associated with Williams Beuren syndrome, the 17p11.2 deletion associated with Smith-Magenis Syndrome, and the reciprocal duplication, associated with Potocki-Lupski Syndrome, among others[25].
In this context, it is very important that the whole family (or at least the mother/father/patient trio) is analyzed, therefore providing direct evidence of family segregation and a straightforward filtering of the variants observed in the proband, in order to yield a proper molecular diagnosis and make interpretation of variants easier.
Functional understanding of ID-related genetic variants
Once a new genetic variant is identified, understanding its relationship with the biological molecular mechanism is the next important step. Concomitant with the explosion of genomic information came a revolution of tools that enabled the genetic modification of genomes. CRISPR/Cas and its associated technologies are versatiles and make gene and genomic editing much easier than before. Model organisms have been very helpful for studying the effect of a single genetic modification at the level of the organism. Despite the tremendous complexity of ID in humans, it is possible to look for conservation and relevant phenotypes to comprehend the ID-related pathophysiology in model organisms. Hence, now more than ever, model organism studies have become instrumental for understanding the molecular mechanisms underlying ID[26]. This includes mice, which have historically been used to learn about disease biology and to find potential therapeutic strategies, and fruit flies and zebrafish, which have been introduced as disease models for ID as well.
Several extremely useful tools already exist to assess basic processes that inform on gene function, associate a particular locus with ID, and enable dissection of both functional variant types and combinations of variants (biallelic or multilocus) with ID. When a novel genetic variant is identified in a patient, it is very important to define whether the variant is within a known coding region or elsewhere in the genome for this is fundamental to determining future steps [Figure 1]. If the variant is located in a coding region, the next big question is whether it is located within a gene already related to ID If it is a new candidate gene, many different types of evidence can be used to identify functionally associated ID genes. This “guilt by association” concept predicts that if two gene products work in the same pathway or process, then mutations in these genes probably have overlapping phenotypic consequences[27]. For example, genes that encode physically interacting proteins, which are co-regulated or co-evolving, are more likely to work in a common process. In addition, studies on single gene mouse models of ID reveal that the effects of these mutations converge onto similar or related etiological pathways, highlighting common pathological nodes that can help in the understanding of new ID related genes[28]. The huge collection of mutant model organisms and the literature can be reviewed to study ID related phenotypes, keeping in mind the mode of inheritance demonstrated in humans when choosing the model to study. Towards this end, existing mutant mice collections such as the International Mouse Phenotyping Consortium (IMPC, http://www.mousephenotype.org/), the Mouse Genome Informatics (MGI, http://www.informatics.jax.org/) online database, and the European Mouse Mutant Cell Repository (EuMMCR, https://www.eummcr.org/), are all very useful resources that have combined with easy to implement genetic modification tools[29] which are instrumental for rapid understanding of the relationship between a gene and ID.
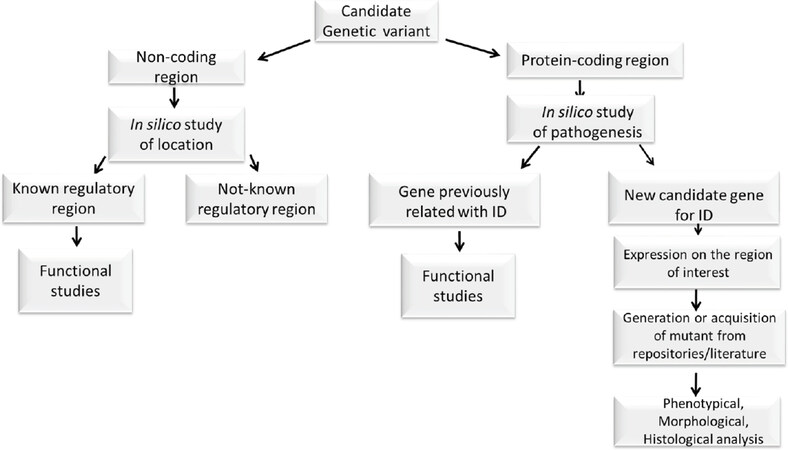
Figure 1. Schematic summary of the understanding of functional consequences of ID-related genetic variant. ID: intellectual disability
If the genetic variant is within a known ID-related gene, it is important to understand its functional relationship with the phenotype. Towards this end, the construction of gene deletion collections in yeasts[30,31] and Escherichia coli[32], the genome-wide RNA interference screens in worms[33] and flies[34] and the availability of mutants in zebrafish, in which partial to full rescue of a zebrafish phenotype by injecting the human orthologous mRNA can be observed[35], all allow quick functional screening. In addition, induced pluripotent stem cells can be used to study rare genetic variants in the complex human genome, as long as the clonal nature of cellular reprogramming and positive selection are well accounted for.
Pathogenic CNVs are significantly enriched for genes involved in development[36] and are particularly increased in neurodevelopmental disorders. Molecular studies of pathogenic CNVs are thus very relevant to ID research. However, pathogenic CNVs are usually very large and contain several physically linked genes. Thus, understanding the cause of ID pathogenicity remains a major challenge although animal models can be very useful towards this goal. Examples include the Smith-Magenis syndrome (SMS, OMIM #182290), associated with a deletion within band p11.2 of chromosome 17, and Potocki-Lupski syndrome (PTLS, OMIM #610883) related with reciprocal duplication. Both syndromes include ID among their clinical presentation. Modeling this pathogenic CNV in mice was possible due to the confirmation of a syntenic genomic region in mice[37] followed by the creation of the desired rearrangement by chromosomal engineering[38,39]. Phenotypic characterization of the resulting mice[40], identification of the responsible gene within the genetic interval[41,42], and analysis of the contribution of the genomic structural change per-se to the ultimate phenotype[43] were all possible with the genetically modified animals. With advancement in technology, efficient and rapid generation of large genomic variants in mice can be achieved in less time than before. For example, the CRISpr MEdiated REarrangement (CRISMERE) strategy. deletions, duplications, and inversions of genomic regions as large as 24.4 Mb can be obtained[44,45], making such studies easier than before.
If the identified genetic variant falls within a non-coding region, the challenge to understand its functional consequence is even greater. Accurate classification of regulatory regions can be of immense help in predicting the biological effects of noncoding genetic variants associated with particular traits and diseases. However, determining whether a given genetic variant affects the function of a regulatory element is still nontrivial. One example is seen with the transcription factor-encoding gene ARX in which protein-coding mutations cause various forms of ID and epilepsy. In contrast, variations in ARX surrounding non-coding sequences are correlated with milder forms of non-syndromic ID and autism. Using zebrafish transgenesis, long-range regulatory domains and brain region-specific enhancers were identified that explained the neuronal phenotypes related to the associated neuropsychiatric disease[46].
From genes to biological pathways
With all these efforts, the biological processes involved in ID are starting to unravel. Genes related to ID are involved in a variety of biological functions and clusters in processes such as metabolism, transporters, nervous system development, RNA metabolism, and transcription[22]. Examples of these functional nodes are discussed next.
The RAS-MAPK (mitogen-activating protein kinase) and the PI3K-AKT-mTOR pathways were first associated with cancer, but are known to be critical for synaptic plasticity and behavior[47]. The RAS-MAPK signaling cascade is a metabolic pathway that regulates growth factors and embryological development and is now associated with syndromic ID such as Noonan (OMIM #163950) and Costello syndromes (OMIM #218040)[48]. ThePI3K-AKT-mTOR signaling cascade contributes by mediating various cellular processes including cell proliferation and growth, and nutrient uptake. Dysregulation of this node has been identified as a cause of several neurodevelopmental diseases, including megaloencephaly, microcephaly, autism spectrum disorder, ID, schizophrenia and epilepsy[49,50].
The RHO-GTPase signaling cascade is associated with a variety of cellular functions including the morphogenesis of dendritic spines. Mutations in both regulators and effectors of the RHO GTPases (i.e., GDI, PAK3, ARHGEF6) have been found to underlie various forms of non-syndromic ID[51]. Mutations in one of the downstream effectors, the calcium/calmodulin-dependent protein kinase type II (CaMKII), have been reported in patients with ID[52]. Moreover, mutations in the cytosolic protein SYNGAP1 (SYNaptic GTPase activating protein) result in a neurodevelopmental disorder termed Mental retardation-type 5 (MRD5, OMIM #612621) with a phenotype consisting of ID, motor impairments, and epilepsy. SYNGAP1 plays critical roles in synaptic development, structure, function, and plasticity and is one of the targets of phosphorylation by CaMKII[53]. This example serves to illustrate the power of identifying pathways towards understanding ID biology.
Pathway convergence[54-57] could stem from the fact that the repertoire of cells affected by ID is limited and therefore, the pathways into which ID-associated variants congregate is a reflection of the specialized function of brain cells. However, the accurate identification of such converging pathways has the potential to help understand brain dysfunction and pathology.
ID associated with epigenetic misregulation
A critical feature of the human brain that underlies cognition and the development of intellectual abilities is the capacity of the nervous system to reorganize its connections functionally and structurally in response to intra- and extra cellular (environmental) clues. This experience-dependent neural plasticity is particularly high during development[58]. Therefore, it is not surprising that in addition to genetic factors, the environment has particular influence during gestation or the early postnatal period and both contribute to the development of ID. Examples of such environmental factors contributing to ID include cerebrovascular incidents associated with premature birth or perinatal asphyxia, prenatal exposure to neurodevelopmental toxins or bacterial and viral infections, maternal conditions such as diabetes, phenylketonuria and immune system alterations, malnutrition (of both mother and child) and specific deficiencies such as that of iodine. Some of these ID-contributing environmental factors affect normal neurodevelopment directly by inducing genetic mutations, enhancing cell death, inhibiting differentiation processes and blocking the activity of key developmental proteins. However, the effects of the vast majority of environmental factors involve gene-environment interactions that drive long-lasting neural and behavioral changes. Currently, these effects are strongly linked with epigenetic changes elicited by environmental factors. For example, emerging evidence suggests that environmental perturbations can alter DNA methylation patterns in the developing brain[59], leading to the currently prevailing theory that changes in the brain methylome likely contribute to the pathogenesis of ID.
Aberrant DNA methylation (induced by environmental factors, stochastically arisen or resulting from an underlying change in DNA sequence) that leads to dysregulated genome function, affecting genes relevant for neurodevelopment and brain plasticity can potentially cause ID. These genomic (epi) variations are missed by conventional sequencing approaches and can potentially underlie a considerable fraction of genetically undiagnosed ID cases. Recently, array-based methylation profiling of a large cohort of patients with neurodevelopmental disorders identified rare epigenetic changes in ~20% of patients[60]. These changes were absent in thousands of controls, repeatedly identified in unrelated patients and located in promoters of known NDD genes, suggesting that abnormal methylation contributes to the phenotype of the patients. Further support for this hypothesis came from findings that epivariations in gene promoters were often associated with changes in gene expression, some of which were so extreme as to mimic the loss of function coding mutations. Thus, the search for epivariations should be considered as a complementary, molecular diagnostic tool in patients with genetically unexplained ID[61].
Consistent with epigenetic mechanisms underlying the development of the brain and cognitive phenotypes, amongst the hundreds of genes already identified as contributing to ID, a large number of them encode for epigenetic regulators[62-65]. Neurodevelopmental disorders exhibiting ID such as Rett syndrome (OMIM #312750 ), ATR-X (OMIM #301040), Kleefstra syndrome (OMIM #610253), Fragile X (OMIM #300624), X-linked syndromic ID (XLID) Claes-Jensen type (OMIM #300534) and Rubinstein-Taybi syndrome (OMIM #180849) are caused by mutations in chromatin-remodeling proteins (ATRX in ATR-X), transcriptional regulators (MeCP2 in Rett syndrome and CREBBP/CBP in Rubinstein-Taybi syndrome) and histone modifiers (EHMT1 in Kleefstra syndrome and KDM5C in XLID). Further, mutations in the DNA methyltransferase gene DNMT3A and HIST1H1E, encoding histone H1.4 were shown to cause ID[66,67].
In summary, generating genotype-phenotype correlations for ID is incredibly complex. This is due in part to the confounding effect of phenotypic and etiologic heterogeneity, along with the rare and variable penetrant nature of the underlying risk variants identified so far[68]. One consequence of this complexity is the application of artificial intelligence (AI) for precision medicine in neurodevelopmental disorders, including ID, autism spectrum disorder and epilepsy, which is still far from accurate. Larger sample sizes and broader (in terms of technologies) studies are expected to allow identification of the relative contributions of each gene/loci to different, but overlapping and highly correlated phenotypes related to ID, such as intelligence quotient (IQ), educational attainment, schizophrenia and depression among others. Finally, increasing data availability will also allow for the development of phenotype specific polygenic risk scores (PRS)[69].
Conclusion
Regardless of the progress made so far, the overall picture is still highly complex and there are plenty of future challenges to be addressed for ID. Is ID a single entity amenable to the application of standard genetic analysis methodologies? Are genetic variants and environmental influences responsible for ID also involved in the normal distribution of IQ? Which of the identified variants are responsible for the final phenotype? What are the contributions of single genes versus that of the genomic makeup? Are the variant effects constitutive, or do they appear only in response to specific environmental challenges? How do we understand the epigenetic contribution to ID? And what are the biological nodes that are promising for therapeutic options? With the amount of genetic information already available, it is clear that the level of complexity in ID is immense and there is an entire genome to investigate and understand. Stratification and careful consideration of ID grouping is also a must. We expect future research strategies to involve the development of animal models and/or in vitro molecular functional studies which will provide reliable, accessible and cost-effective platforms to perform functional tests of novel variants, and accelerate discovery of the biological functions underlying genetic forms of ID and enhance the translation to clinical care.
Declarations
AcknowledgmentsWe thank Erna M. L. Stoddart for English proofreading and formatting assistance.
Authors’ contributionsManuscript writing: Marti M, Millan MIP, Young JI
Conception, design and manuscript writing: Walz K
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Deary IJ, Johnson W, Houlihan LM. Genetic foundations of human intelligence. Hum Genet 2009;126:215-32.
2. Davies G, Tenesa A, Payton A, Yang J, Harris SE, et al. Genome-wide association studies establish that human intelligence is highly heritable and polygenic. Mol Psychiatry 2011;16:996-1005.
3. Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, et al. Strong association of de novo copy number mutations with autism. Science 2007;316:445-9.
4. Gonzalez-Mantilla AJ, Moreno-De-Luca A, Ledbetter DH, Martin CL. A cross-disorder method to identify novel candidate genes for developmental brain disorders. JAMA Psychiatry 2016;73:275-83.
5. Tasse MJ, Luckasson R, Schalock RL. The relation between intellectual functioning and adaptive behavior in the diagnosis of intellectual disability. Intellect Dev Disabil 2016;54:381-90.
6. Dalsgaard S, Thorsteinsson E, Trabjerg BB, Schullehner J, Plana-Ripoll O, et al. Incidence rates and cumulative incidences of the full spectrum of diagnosed mental disorders in childhood and adolescence. JAMA Psychiatry 2019;77:155-64.
7. Boyle CA, Boulet S, Schieve LA, Cohen RA, Blumberg SJ, et al. Trends in the prevalence of developmental disabilities in US children, 1997-2008. Pediatrics 2011;127:1034-42.
8. Van Naarden Braun K, Christensen D, Doernberg N, Schieve L, Rice C, et al. Trends in the prevalence of autism spectrum disorder, cerebral palsy, hearing loss, intellectual disability, and vision impairment, metropolitan atlanta, 1991-2010. PLoS One 2015;10:e0124120.
9. Willemsen MH, Kleefstra T. Genetic diagnostics in intellectual disability: what is the benefit? Ned Tijdschr Geneeskd 2014;158:A8098.
10. Blesson A, Cohen JS. Genetic counseling in neurodevelopmental disorders. Cold Spring Harb Perspect Med 2020;10:a036533.
11. Posthuma D, Polderman TJ. What have we learned from recent twin studies about the etiology of neurodevelopmental disorders? Curr Opin Neurol 2013;26:111-21.
12. Shendure J, Findlay GM, Snyder MW. Genomic medicine-progress, pitfalls, and promise. Cell 2019;177:45-57.
13. Coulter ME, Miller DT, Harris DJ, Hawley P, Picker J, et al. Chromosomal microarray testing influences medical management. Genet Med 2011;13:770-6.
14. Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014;515:216-21.
15. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749-64.
16. de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921-9.
17. Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014;511:344-7.
18. Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014;312:1880-7.
19. Stessman HA, Xiong B, Coe BP, Wang T, Hoekzema K, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet 2017;49:515-26.
20. Jiang YH, Yuen RK, Jin X, Wang M, Chen N, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet 2013;93:249-63.
21. Johns Hopkins University. OMIM® - Online Mendelian Inheritance in Man®. Available from: https://www.omim.org/search/?index=entry&search=intellectual+disability&sort=chromosome_number+asc%2C+chromosome_sort+asc&start=1&limit=100&retrieve=geneMap&gm_exists=true [Last accessed on 17 Apr 2020].
22. Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, et al. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet 2016;98:149-64.
23. Richards S, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.
24. Chen C, Chen D, Xue H, Liu X, Zhang T, et al. IDGenetics: a comprehensive database for genes and mutations of intellectual disability related disorders. Neurosci Lett 2018;685:96-101.
25. Morrow EM. Genomic copy number variation in disorders of cognitive development. J Am Acad Child Adolesc Psychiatry 2010;49:1091-104.
26. In: Walz K, Young JI, editors. Cellular and animal models in human genomics research. Translational and applied genomics series. San Diego: Elsevier/Academic Press; 2019. pp. 1-226.
28. Verma V, Paul A, Amrapali Vishwanath A, Vaidya B, Clement JP. Understanding intellectual disability and autism spectrum disorders from common mouse models: synapses to behaviour. Open Biol 2019;9:180265.
29. Canales CP, Walz K. The mouse, a model organism for biomedical research. In: Walz K, Young JI, editors. Cellular and animal models in human genomics research. Translational and Applied Genomics Series. San Diego: Elsevier/Academic Press; 2019. pp. 119-40.
30. Giaever G, Chu AM, Ni L, Connelly C, Riles L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002;418:387-91.
31. Kim DU, Hayles J, Kim D, Wood V, Park HO, et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol 2010;28:617-23.
32. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2006;2:2006.0008.
33. Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003;421:231-7.
34. Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007;448:151-6.
35. Lepanto P, Zolesi FR, Badano JL. Studying human genetic variation in zebrafish. In: Walz K, Young JI, editors. Cellular and animal models in human genomics research. Translational and Applied Genomics Series. In: Anonymous San Diego: Elsevier/Academic Press; 2019. pp. 89-117.
36. Rice AM, McLysaght A. Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat Commun 2017;8:14366.
37. Bi W, Yan J, Stankiewicz P, Park SS, Walz K, et al. Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. Genome Res 2002;12:713-28.
39. Zheng B, Mills AA, Bradley A. A system for rapid generation of coat color-tagged knockouts and defined chromosomal rearrangements in mice. Nucleic Acids Res 1999;27:2354-60.
40. Walz K, Caratini-Rivera S, Bi W, Fonseca P, Mansouri DL, et al. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance. Mol Cell Biol 2003;23:3646-55.
41. Bi W, Ohyama T, Nakamura H, Yan J, Visvanathan J, et al. Inactivation of Rai1 in mice recapitulates phenotypes observed in chromosome engineered mouse models for Smith-Magenis syndrome. Hum Mol Genet 2005;14:983-95.
42. Walz K, Paylor R, Yan J, Bi W, Lupski JR. Rai1 duplication causes physical and behavioral phenotypes in a mouse model of dup(17) (p11.2p11.2). J Clin Invest 2006;116:3035-41.
43. Ricard G, Molina J, Chrast J, Gu W, Gheldof N, et al. Phenotypic consequences of copy number variation: insights from Smith-Magenis and Potocki-Lupski syndrome mouse models. PLoS Biol 2010;8:e1000543.
44. Birling MC, Schaeffer L, Andre P, Lindner L, Marechal D, et al. Efficient and rapid generation of large genomic variants in rats and mice using CRISMERE. Sci Rep 2017;7:43331.
45. Birling MC, Herault Y, Pavlovic G. Modeling human disease in rodents by CRISPR/Cas9 genome editing. Mamm Genome 2017;28:291-301.
46. Ishibashi M, Manning E, Shoubridge C, Krecsmarik M, Hawkins TA, et al. Copy number variants in patients with intellectual disability affect the regulation of ARX transcription factor gene. Hum Genet 2015;134:1163-82.
47. Borrie SC, Brems H, Legius E, Bagni C. Cognitive dysfunctions in intellectual disabilities: the contributions of the ras-MAPK and PI3K-AKT-mTOR pathways. Annu Rev Genomics Hum Genet 2017;18:115-42.
48. Schubbert S, Bollag G, Shannon K. Deregulated Ras signaling in developmental disorders: new tricks for an old dog. Curr Opin Genet Dev 2007;17:15-22.
49. Wang L, Zhou K, Fu Z, Yu D, Huang H, et al. Brain development and akt signaling: the crossroads of signaling pathway and neurodevelopmental diseases. J Mol Neurosci 2017;61:379-84.
50. Dobyns WB, Mirzaa GM. Megalencephaly syndromes associated with mutations of core components of the PI3K-AKT-MTOR pathway: PIK3CA, PIK3R2, AKT3, and MTOR. Am J Med Genet C Semin Med Genet 2019;181:582-90.
51. Ba W, van der Raadt J, Nadif Kasri N. Rho GTPase signaling at the synapse: implications for intellectual disability. Exp Cell Res 2013;319:2368-74.
52. Chia PH, Zhong FL, Niwa S, Bonnard C, Utami KH, et al. A homozygous loss-of-function CAMK2A mutation causes growth delay, frequent seizures and severe intellectual disability. Elife 2018;7:e32451.
53. Agarwal M, Johnston MV, Stafstrom CE. SYNGAP1 mutations: clinical, genetic, and pathophysiological features. Int J Dev Neurosci 2019;78:65-76.
54. Chen ES, Gigek CO, Rosenfeld JA, Diallo AB, Maussion G, et al. Molecular convergence of neurodevelopmental disorders. Am J Hum Genet 2014;95:490-508.
55. Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018;359:693-7.
56. Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011;474:380-4.
57. Frega M, Selten M, Mossink B, Keller JM, Linda K, et al. Distinct pathogenic genes causing intellectual disability and autism exhibit a common neuronal network hyperactivity phenotype. Cell Rep 2020;30:173-86.e6.
58. Gallegos DA, Chan U, Chen LF, West AE. Chromatin regulation of neuronal maturation and plasticity. Trends Neurosci 2018;41:311-24.
59. Keil KP, Lein PJ. DNA methylation: a mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ Epigenet 2016;2:dvv01210.
60. Barbosa M, Joshi RS, Garg P, Martin-Trujillo A, Patel N, et al. Identification of rare de novo epigenetic variations in congenital disorders. Nat Commun 2018;9:2064.
61. Aref-Eshghi E, Bend EG, Colaiacovo S, Caudle M, Chakrabarti R, et al. Diagnostic utility of genome-wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am J Hum Genet 2019;104:685-700.
62. Kleefstra T, Schenck A, Kramer JM, van Bokhoven H. The genetics of cognitive epigenetics. Neuropharmacology 2014;80:83-94.
64. Gabriele M, Lopez Tobon A, D’Agostino G, Testa G. The chromatin basis of neurodevelopmental disorders: Rethinking dysfunction along the molecular and temporal axes. Prog Neuropsychopharmacol Biol Psychiatry 2018;84:306-27.
65. Fahrner JA, Bjornsson HT. Mendelian disorders of the epigenetic machinery: postnatal malleability and therapeutic prospects. Hum Mol Genet 2019;28:R254-64.
66. Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 2014;46:385-8.
67. Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, et al. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet 2017;100:725-36.
68. Uddin M, Wang Y, Woodbury-Smith M. Artificial intelligence for precision medicine in neurodevelopmental disorders. NPJ Digit Med 2019;2:112.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Marti M, Millan MIP, Young JI, Walz K. Intellectual disability, the long way from genes to biological mechanisms. J Transl Genet Genom 2020;4:104-13. http://dx.doi.org/10.20517/jtgg.2020.10
AMA Style
Marti M, Millan MIP, Young JI, Walz K. Intellectual disability, the long way from genes to biological mechanisms. Journal of Translational Genetics and Genomics. 2020; 4(2): 104-13. http://dx.doi.org/10.20517/jtgg.2020.10
Chicago/Turabian Style
Marti, Marcelo, Maria Ines Perez Millan, Juan I. Young, Katherina Walz. 2020. "Intellectual disability, the long way from genes to biological mechanisms" Journal of Translational Genetics and Genomics. 4, no.2: 104-13. http://dx.doi.org/10.20517/jtgg.2020.10
ACS Style
Marti, M.; Millan MIP.; Young JI.; Walz K. Intellectual disability, the long way from genes to biological mechanisms. J. Transl. Genet. Genom. 2020, 4, 104-13. http://dx.doi.org/10.20517/jtgg.2020.10
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 25 clicks
Cite This Article 25 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.