
Cryogenic electron paramagnetic resonance spectroscopy of flash-frozen tissue for characterization of mitochondrial disease
 0
0Abstract
Electron paramagnetic resonance spectroscopy (EPR) is an analytical technique that, uniquely, can be used to directly interrogate flash-frozen tissue. Quantitative information on the thermodynamic potential of the mitochondrion to synthesize ATP, and the extent of reactive oxygen species-mediated oxidative stress on the mitochondrion and the cell at large, can be obtained. A compromised ability to synthesize ATP and oxidative stress are two of the characteristic sequelae of mitochondrial disease and therapeutic approaches may differ widely depending on which of these is dominant. EPR, therefore, has a role to play in the characterization, diagnosis, and ongoing evaluation of therapies for mitochondrial disease in human patients and model systems. An introduction to mitochondrial disease is followed by a description of EPR, a summary of the EPR signals that can be expected from tissue samples, sample preparation and analytical methods, and a case study in which EPR and complementary techniques were employed on a rat model to study human mitochondrial disease.
Keywords
Introduction
Mitochondria are characterized by mitochondrial respiratory chain (MRC) complexes that catalyze redox reactions and act as electron transfer conduits during energy metabolism, driving ATP synthesis while closely chaperoning potentially toxic one-electron redox equivalents. The MRC proteins contain a variety of redox-active centers, including iron-sulfur clusters, heme, copper ions, and quinones. Each of these can exist in distinct oxidation states with environmental dependencies that include (1) the redox environment of the mitochondrion and the cell; (2) the oxidative stress burden and history; (3) the integrity of the mitochondrial membrane; and (4) the functionality of the individual MRC components and the electron transfer chain overall. Other non-MRC metalloproteins, particularly aconitase and catalase, provide distinct and specific biomarkers for oxidative stress.
Mitochondrial diseases (MD) can arise where depletion of mitochondrial DNA (mtDNA)[1,2], or mutations in mtDNA and/or nuclear DNA lead to altered mitochondrial function[3-7]. Altered catalytic and electron transferring activities of mitochondrial complexes I-V have been associated with MD, and physiological consequences of MRC defects include reduced metabolic capacity, reduced ATP synthesis, and increased oxidative and nitrosative stress[8-18]. Symptoms are manifold and include weakness (from central nervous system, peripheral nerve, and/or skeletal muscle disease), pain, intolerance of some general anesthetics and anti-epileptic drugs, gastrointestinal disorders, ophthalmoplegia and/or visual failure, failure to thrive, cardiac and respiratory disease, liver disease, diabetes, seizures, sensorineural hearing loss, mental retardation, dementia, movement disorders, increased susceptibility to infection, and pregnancy loss[5,6,19-41]. The primary manifestation of mitochondrial dysfunction is an inability to generate enough energy from metabolism to maintain the functional or structural integrity of the associated tissues. Thus, MD is particularly debilitating when dysfunctional mitochondria are present in tissues with high energy requirements, such as the cerebrum, nerves and muscle. The other major underlying pathology is oxidative stress i.e. the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS and RNS are free radicals and related compounds, some of which exhibit high reactivity and can damage proteins, lipids and nucleic acids. Depolarization of the mitochondrial membrane can occur due to physical damage. Damage to individual MRC components can exacerbate oxidative stress and an oxidative stress cascade can occur. Identification of low ATP production or oxidative stress to be primarily responsible for symptoms may have profound consequences for subsequent care and therapy.
Traditional diagnosis of MD includes clinical presentation of symptoms, family history, pathology, metabolic profiling, enzyme activity levels, electrophysiology, magnetic resonance imaging of brain and magnetic resonance spectroscopy of metabolites, and mtDNA analysis[7,10,34,42-55]. Diagnosis can be challenging, given that (1) mitochondrial metabolism can be affected in non-mitochondrial diseases; (2) there can be extensive variability in the distribution of abnormal mitochondria within an individual patient, resulting in “false negative” testing to occur when tissues containing the abnormal mitochondria are not tested; and (3) there are no uniform, clear cut pathological abnormalities to distinguish all MD patients from patients with other disorders, to the extent that some biopsy specimens look structurally normal. MD can also present with an extraordinary range of clinical symptoms, and laboratory testing abnormalities are common. MD is often suspected clinically as part of the differential diagnosis in patients with diseases involving the brain, muscle, or liver and MD-like symptoms are often exhibited in early childhood. Attempts to improve MD diagnosis have included the use of diagnostic algorithms to predict the likelihood of MD, DNA sequencing, and omics methods, each with associated advantages and challenges of their own[49,56-62].
Herein, the application of cryogenic electron paramagnetic resonance spectroscopy (EPR) of intact, flash-frozen tissue is described for the diagnosis and characterization of metabolic dysfunction in general, and MD in particular. The article describes (1) the principles of EPR; (2) the EPR signals exhibited by mammalian and human tissue; (3) sample preparation considerations; (4) analysis methods; and (5) the relevance of EPR to MD and integration of EPR results with other, complementary investigative methods.
Electron paramagnetic resonance
EPR, in the present context, is the measurement of the magnetic field-dependence of absorption of a photon by a paramagnetic substance i.e. containing unpaired electrons in one or more atomic, ionic, or molecular orbitals with a non-zero net spin-magnetic moment[63]. Almost all paramagnets of biomedical importance are either free radicals, transition metal ions, or clusters. The magnetic field-dependent resonant spin-transition of a paramagnetic electron occurs via its interaction with the oscillating magnetic field of an incident photon. The total “magnetic field” experienced by an electron can be due to (1) contributions from an applied laboratory magnetic field; (2) arising from spin-orbit coupling; (3) arising from zero-field splitting in individual ions or radicals containing more than one unpaired electron; (4) from nearby additional unpaired electrons (exchange- and dipolar-couplings); and (5) from nearby magnetic nuclei (electron-nuclear hyperfine coupling)[64]. Additional “fields” due to nuclear Zeeman and quadrupolar interactions can generally be neglected in the present context. EPR can, in principle, provide a wealth of information on the identity, chemical nature, chemical environment, electronic structure, and physical structure of the analyte from analysis of each of these interactions, typically employing computer analysis and simulations, and increasingly with quantum chemistry calculations (density functional theory, Taylor theory)[65-76]. However, in the case of biological tissues, the origins and spectroscopic parameters of many of the EPR signals have been well-characterized, as described in detail below. In the studies of concern here, one is primarily interested in (1) assigning each of the signals; and (2) quantifying the species responsible.
Experimentally, the sample is placed in a resonant structure that supports a standing microwave of fixed frequency, and the applied magnetic field is scanned to search for resonant absorption across a wide field envelope[77]. A typical microwave frequency is 9.5 GHz, with magnetic field scans of 0-1 T (0-10,000 G; the derived unit of magnetic flux density, the gauss G, is generally used to label the abscissae of EPR spectra). Multiple conflicting factors need to be considered when choosing a frequency for EPR. The present author’s opinion is that 18 GHz may be the best overall frequency for studies on biological tissue and commercial, high-quality 9.5 GHz instruments represent a reasonable compromise. Higher frequencies correspondingly require higher fields and superconducting magnets are generally needed for EPR at > 35 GHz. The EPR spectrum is often presented in the derivative-like mode, ∂χ”/∂B0, due to the use of magnetic field modulation and phase-sensitive detection, and resonant lines in the EPR spectrum are often labeled with “geff-values” in order to remove the frequency-dependence from the resonance position label, as geff = hn/bB, where n is the microwave frequency and B is the resonant field. EPR spectra recorded at different frequencies (e.g., on different instruments) can thus be compared using the geff-values of the signals. Where S = 1/2, geff is equal to g, the Landé g-factor incorporating the Zeeman and spin-orbit coupling terms; where S > 1/2, geff is related to g through the zero-field splitting term E/D[69]. Up to three geff -values for any Kramers’ doublet may be observed, corresponding to the principal tensor orientations xx, yy, and zz, and are often labeled gx, gy, gz, where relationships to molecular or electronic structure symmetry are obvious, or g1, g2, g3, otherwise. Notably, the value of g for the free electron, and many S = 1/2 systems, is close to 2, whereas for S > 1/2 the highest value, geff max ≤ 4S.
EPR analysis of tissue samples for studies of MD must be carried out at low temperatures. Trivially, but nevertheless important, tissues must be maintained at a temperature sufficiently low (e.g., -80 °C freezer or liquid nitrogen at 77 K) to prevent molecular diffusion that will change the signals over time, so a low temperature is necessary for data collection to preserve sample integrity. Second, to a crude approximation, available EPR signal intensity is often inversely proportional to the absolute temperature, and signal-to-noise can be limited with raw biological material so maximizing it is important. Third, and more fundamentally, substantial population of the ground state in spin-systems with more than one unpaired electron (S > 1/2) may require very low temperatures, close to liquid helium (4.2 K). Knowledge of the population of the ground state is necessary for quantitation of signals. The final and often limiting determinant of the temperature for EPR data collection is the relaxation kinetics of the EPR signals[78]; these vary considerably among the signals observed from biological tissues. Although the temperature can be tailored for specific investigation of an individual signal, there is no one optimum temperature for global EPR analysis of biological tissue analysis; however, data collection at 2 temperatures, 12 K and 40 K, is often sufficient when combined with careful analysis[79].
EPR signals from frozen tissue
The low-temperature EPR signals from tissues including liver, muscle, heart, and brain, have largely been assigned from comparison of signals from tissues with those from fractionated extracts, purified proteins or protein complexes, modified or truncated proteins or protein complexes, and isolated mitochondria[80-82]. The predominant signals are those from the MRC complexes I-IV, with additional signals from semiquinones, aconitase, catalase, and from ferriheme and transferrin in residual blood. The centers from MRC complexes and their computer-simulated EPR signals are shown in Figure 1.
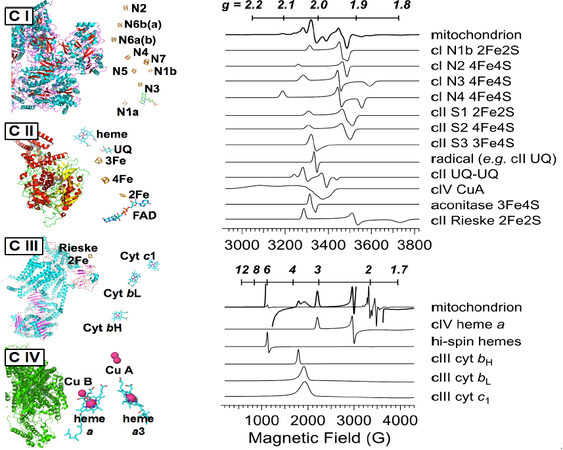
Figure 1. Redox centers in the mitochondrial respiratory chain (left) and EPR signals observed in frozen tissue with geff -values on the top scale and resonant fields at 9.5 GHz on the bottom scale (right). The labels “C I”, “C II”, “C III”, and “C IV” refer to mitochondrial respiratory chain complexes I, II, III, and IV, respectively. The spectra are computer simulations of the signals as expressed in whole, unprocessed, frozen tissue; details of the signals (precise geff values; splittings; line widths) can differ noticeably in isolated proteins or mitochondrial fragments, particularly due to differences in redox potential and spin-spin interactions between redox centers. The signal due to the whole mitochondrion in the lower right panel is shown at two different amplitudes superimposed with one at 20 × the amplitude of the other, in order to have appreciable amplitude of each of the contributory signals in one or other of the traces (this is clearest at geff = 6), while the signals shown individually have normalized peak amplitudes
Of the MRC complexes, Complex I (NADH: quinone oxidoreductase) provides the richest array of EPR signals due to the large number of [2Fe2S] and [4Fe4S] clusters that exhibit EPR signals in the monocationic reduced state, of which seven are proposed to be integral to electron transfer through the complex, based on the bacterial enzyme[83]. There has, however, been some controversy over the assignment of EPR signals of FeS clusters in mammalian Complex I to structurally characterized ones because of the extensive overlap of signals from both Complex I and other sources in the spectra of intact tissue, and complicating weak magnetic interactions between the clusters in the intact holoprotein in situ[84-86]. The most informative [FeS] clusters in Complex I are, thankfully, also the best-resolved in the EPR spectrum, N1b, N2, N3, and N4[79,83]. Consideration of only these clusters reduces the number of computer-fitting parameters to avoid over-fitting to a large number of highly correlated parameters but nevertheless, provides a useful interrogation of the redox status of the mitochondrion in the low-potential (reducing) regime. Two of the clusters, the [4Fe4S]+ N4 (-280 mV) and N3 (-325 mV), exhibit well-resolved g3 resonances at g = 1.88 and 1.86, respectively. These clusters exhibit the lowest midpoint potentials that can be accessed by an NADH+/NAD couple-determined overall redox potential and high intensities of signals from these centers indicate a highly reducing environment. The N1b [2Fe2S]+ and N2 [4Fe4S]+ clusters have higher midpoint potentials, around -205 mV to -270 mV, depending on the overall redox status of other clusters, and exhibit essentially axial EPR spectra with g⟂ ~ 1.92.
The signal from Complex II (succinate dehydrogenase) is dominated by two overlapping signals due to reduced [2Fe2S]+ (S1) and [4Fe4S]+ (S2) clusters. The precise g-values for these signals are dependent on the extent of reduction and the temperature, as the influence of the spin-spin interaction between them is dependent on both. However, they both give rise to an intense derivative feature at g ~1.92, adding to the contribution from Complex I signals[87-89]. A third complex II signal due to an oxidized [3Fe4S]+ (S3) signal overlaps at g ~2.02, with that from oxidatively-deactivated cytosolic aconitase in which the labile Fea atom is lost from the active, EPR-silent [4Fe4S]2+ cluster to form an [3Fe4S]+ cluster[90]. However, the very different temperature dependences of S3 and aconitase allow deconvolution by recording at 2 temperatures (e.g., 12 and 40 K)[80]. Another signal in that region with a g ~2.015 turning point and flanking resonances at g ~2.03 and 1.98 has been assigned to a stable, dipolar-coupled ubisemiquinone pair in the vicinity of S3 in Complex II[91].
Although the redox cofactors of Complex III (cytochrome bc1 complex; CoQH2-cyctochrome c reductase) have been studied extensively in vitro, the EPR spectra of tissue and cells do not provide much information due to their low intensities. The only signal routinely assignable to complex III is the reduced [2Fe2S]+ Rieske cluster with a distinct sharp g1 resonance at 2.03, a derivative feature at 1.90 (generally not resolved from the composite ‘g = 1.92’ signal), and a very broad g3 feature at g ~1.78 that is observable in heart and muscle samples that do not exhibit overlapping Mn(II) signals[79,92-94]. The other EPR signals observable from isolated complex III are resonances that overlap in the region geff ~3.8-3.3 and are due to g1 of cytochromes c1, bL, and bH[92,95]. These signals are sometimes detected but are difficult to characterize and quantify because they are weak, part of a very broad signal envelope, and their resonance positions and line widths can be sensitive to the specific environment.
The final redox-active MRC component, Complex IV (cytochrome c oxidase), exhibits resonances at geff ~3.0 and geff ~2.2 due to low-spin heme a, and, under some conditions, resonances at g ~2.18 and 2.0 due to the dinuclear S = 1/2 CuA center[96,97]. Additional complex IV signals at geff ~12 and 2.95 can be difficult to detect in some tissues and cells due to low signal levels and overlap with the heme ageff ~3.0 resonance, respectively, and are associated with the heme a3-CuB coupled center[98-100].
Additional signals that may be observed in the spectrum arise from the [3Fe4S]+ cluster of aconitase described above[90]; a rhombic high-spin ferriheme signal from catalase with gx ~6.45 and gy ~5.33[81,101], and a signal at g ~4.2, with a characteristic splitting at the crossover, due to transferrin Fe3+[102-104]. A characteristic, and sometimes intense six-line I = 5/2 hyperfine-split signal due the S = 5/2, MS = ±1/2 manifold of 55Mn2+ is observed in some tissues, particularly liver, and may be distorted due to the rapid-passage relaxation effects at the low temperatures needed to observe the other signals[79].
Tissue sample preparation for EPR
The goals of EPR of tissue for the characterization of MD are (1) to provide a snapshot of the redox status of metabolism in actively metabolizing tissue; and (2) to report on instantaneous and chronic exposure to ROS. It is important, then, that tissue is excised and frozen before either the exhaustion of reducing equivalents or of a terminal electron acceptor (usually oxygen) alters the local and global redox potentials, and before further non-physiological ROS-mediated damage occurs. Traditional tissue mounting techniques cannot be used: EPR spectra of formalin-fixed brain tissue, for example[105], are devoid of almost all of the characteristic signals observed in freshly frozen brain[106,107]. Similarly, human tissue-bank muscle samples exhibited intense free-radical signals and signals due to Fe3+ but no signals ascribable to metabolic components were observed. In contrast, studies in the present author’s laboratory indicate that tissue samples frozen between 30 s and 3 min of being harvested from freshly sacrificed animals were rich in signals from MRC redox centers, and the spectra showed no time-dependent changes over that time span. Samples taken from different parts of muscle, liver and lung from the same wild-type (w/t) rat exhibited astounding reproducibility, with signal intensities within 5% for all signals. Between animals, the reproducibility was within 10% (except for the intensities of signals due to blood components, which did vary significantly). Liver samples have been found to be completely stable for at least 6 months at -80 °C. A sample of diced brain that had been stored for 1 year at -80 °C and had experienced substantial sampling handling (during which time it was likely that the temperature rose significantly above -80 °C, though it had never been thawed) showed significant change (+ 10%) in the aconitase region of the signal, indicative of oxidative conversion of the labile [4Fe4S] to [3Fe4S]. Otherwise, only a very minor change (< 5%) in the composite FeS resonance at geff = 1.92 was seen. Liver MRC signals were surprisingly tolerant of freeze-thawing, with only very small changes (< 5%) observed in the aconitase/S3/UQ2 region of the spectrum. Muscle was much less tolerant of freeze-thaw cycles, with large increases in the signals from ferriheme, and aconitase and/or S3, indicating both structural damage and oxidation. Small but significant changes in other signals included an increase in CuA, a decrease in the Complex I/II composite FeS signal at geff = 1.92, and specifically a larger decrease in N3 compared to N4 or the geff = 1.93 feature, suggesting an increase in the overall redox potential. It appears then that medium term storage at -80 °C preserves the EPR signals and thus the redox status of the mitochondria, but sample handling must be carried out carefully to avoid warming, and thawing must be avoided.
Samples are most readily prepared by transfer of fresh tissue into an EPR tube followed by freezing in dry-ice/methanol, liquid nitrogen-chilled isopentane, or liquid nitrogen; the latter is a slower freezing method but is more convenient and safer in a clinical environment due to the lack of flammability. The sample is typically prepared by rapid extrusion of intact tissue from a syringe into an EPR tube that is blown from a length of quartz to leave a small hole in the bottom to prevent air spring resistance. Ideally, samples should completely fill the volume of the tube that occupies the active length of the EPR resonator for reproducible quantitation. Smaller samples can be mounted in the center of the active region of the resonator by first freezing a platform of a water-glycerol mixture at the bottom of the tube, adding the tissue, and adding more water-glycerol to the desired height before freezing. The latter step maintains a more-or-less uniform dielectric constant along the active region of the resonator, which maintains consistency of B1, the oscillating field due to the microwave, across the sample and between samples for a given microwave power and resonator.
Quantitative analysis of EPR spectra
There are a number of challenges to data analysis. One complicating factor is that each of the contributory signals exhibits different dependencies on temperature and microwave power. Therefore, an empirical scaling factor needs to be determined for each signal for any given set of conditions. Phenomena that can reduce the observed intensity of the EPR signal include rapid-passage effects at too-low temperatures, relaxation-broadening at too-high temperatures, and power saturation at too-high microwave power. The most efficient way to arrive at scaling factors is to first determine the temperature dependence of each signal in order to (1) identify a temperature (or a small number of temperatures) at which most or all of the signals are observable; and (2) provide a temperature coefficient to account for any deficiency in signal intensity at the temperatures to be used for routine measurement compared to the maximum intensity that the temperature-dependence experiments predict. Then, a power-dependence at the preferred temperatures is carried out. Modern commercial instruments allow for two-dimensional experiments that can be programmed and run overnight to rapidly facilitate this process.
A second challenge is that many of the signals overlap, sometimes extensively. From the 24 MRC redox centers distributed among Complexes I-IV, 18 discrete EPR signals have been observed from tissues and mammalian cells under various conditions (FeS N1b, N2, N3 & N4 from Complex I; ferriheme, [UQ•]2 and FeS S1, S2 & S3 from Complex II; Rieske FeS and Cyts bL, bH & c1 from Complex III; and CuA, heme a, and a spin-coupled Cu-heme a3 from Complex IV), along with signals from the [3Fe4S]+ of cytosolic and mitochondrial aconitases and free radical species (likely ubisemiquinone, UQ•)[79,82,108-111]. Additional well-characterized tissue-specific signals include Fe(III) from transferrin (Tf), Cu(II) from ceruloplasmin (Cp), high- and low-spin hemes from catalase, and Mn(II). The overlap means that the usual method for quantifying EPR signals, that of double integration and comparison to a standard, with application of the aforementioned scaling factors, is not feasible in most cases. In any case, the wide field envelopes of many of the signals renders integration unreliable, though for some sharp and intense signals, quantitation by integration with scaling factors can be carried out. Instead, the approach taken is to iteratively fit a library of simulated EPR signals [see Figure 1, above] using a Levenberg-Marquardt damped least-squares algorithm (IGORPro, Wavemetrics) [Figure 2][112-114]. The individual computed signals in the library are scaled by the aforementioned empirical scaling factor. While it is unnecessary to determine the scaling factor for each sample, it is important to determine the range of applicability of these factors across the range of samples encountered, particularly as local relaxation parameters are sensitive to the wider redox status. As with all experimentation, the space of controls must be carefully explored.
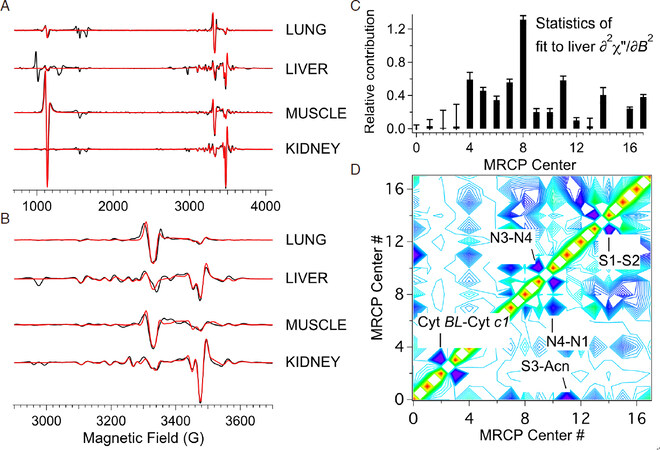
Figure 2. A and B: 9.5 GHz, 12 K, ∂2χ”/∂B02 EPR spectra from flash-frozen tissue samples of w/t rats over the entire spectral region (A) and around geff = 2 (B). Black traces are experimental data and red traces are computer simulations from Levenberg-Marquardt fitting; C: histogram obtained from Levenberg-Marquardt fitting of the liver spectrum to 18 distinct MRC redox centers; D: correlation plot highlighting pairs of redox centers with highly correlated calculated contributions to the overall EPR spectrum. Areas of very high correlation (dark blue) indicate that the fitting can be satisfied by an arbitrary linear combination of the two correlated signals, e.g., for two signals A and B, the fit is satisfied by xA +yB where the value of (x + y) is important but the individual values of x and y are not
The best results have been found by fitting the ∂2χ”/∂B02 “second-derivative” display [compare Figure 1 and Figure 2A and B], though this requires very high quality data that in turn requires ~300 mg of tissue, which is feasible with some animal experiments whereas only 20-50 mg of human biopsy tissue may be available, in which case the fitting is carried out on the raw experimental ∂χ”/∂B0 spectrum. The Levenberg-Marquardt algorithm provides two important parameters of fitting quality. One is a standard deviation that, when compared to the actual contribution of a signal to the to the spectrum, indicates the significance of including that particular signal in the fit [Figure 2C]; a “confidence level” can also be readily calculated. The second is a correlation matrix [Figure 2D] that indicates cases where the calculated contribution of one signal is heavily dependent on the contribution of another. The highly correlated pairs that are seen in tissues are cyt bL-cyt c1; N3-N4; S1-S2; S3-aconitase; and, less so, N1b-N4 and N1b-N2. The N4 and N3 signals each have a small but isolated resonance corresponding to a particular principal orientation and can thus be fitted manually rather than iteratively and constrained thereafter. Cyts bL and c1 are rarely observed with high intensity and signals from either or both can be taken as markers of unusually elevated redox potential. [FeS] clusters S1 and S2 can be deconvoluted with care by exploiting differences in relaxation behavior but this is hardly worth the effort as they essentially provide the same information in almost all cases and are treated together as (S1 + S2); similarly, N1b and N2 can be considered together[84,85]. The most important coupled pair, then, is S3-aconitase, which overlaps extensively across their narrow field envelopes and yet provide very different information that renders their deconvolution important. Fortunately, these are readily distinguished by their very different temperature dependences[80]. Where disease tissue is available with a reliable control, high sensitivity to changes can be accomplished by generating difference spectra (i.e., disease minus control). One could, in principle, merely fit the difference spectrum, in which many of the signals from the individual spectra, those that are not affected by disease, would have cancelled out. A good measure of the effectiveness of the fitting procedure, however, is that the difference spectra of the two individual calculated simulations are indistinguishable from fitting to the difference spectrum directly and provide excellent reproduction of the difference of the experimental spectra[79].
Application of EPR to mitochondrial disease
EPR as a stand-alone technique provides three pieces of information relevant to MD and other diseases and conditions with metabolic components including cancer, neurological diseases, and cardiac dysfunction[79,82,106,108-111,115]. The first is a measure of the redox potential across the MRC by quantitation of signals due to oxidized and reduced redox centers. This is important because the redox potential is the thermodynamic driving force for catalysis and electron transport and, ultimately, represents the potential energy available for conversion to chemical energy via ATP synthesis. The characteristic sharp distinct signal observed at g = 1.92 is largely due to the gx,y features of complex I reduced FeS clusters N1b and N2, with additional contributions from gy of N3 and N4 and from gx,y of complex II S1 and S2. The dominant N1b and N2 clusters exhibit midpoint potentials of -205 mV to -270 mV[84], and diminution of the g = 1.92 signal compared with healthy cells or tissue therefore represents a significant departure of the redox potential from the expected -320 mV, dictated by the NAD+/NADH couple[116]. More sensitive still are the N4 (g3 = 1.88; Em = -280 mV) and N3 (g3 = 1.86; Em = -325 mV) clusters, with the isolated g3 features upfield of the g = 1.92 signal. Care must be taken when Mn2+ is present, particularly prevalent in the liver, as the N3 g3 feature overlaps with the often more-intense high-field MI = -5/2 resonance of the DMS = 1/2 manifold of S = 5/2 Mn2+[79,108]. The absence of the N3 and N4 signals in spectra where the g = 1.92 is well-developed is indicative of either reduced metabolic potential (elevated redox potential), due to inefficient primary metabolism, a compromised MRC, or a membrane that allows reducing equivalents to non-productively drain from the MRC. Another possible cause, though less likely, is specific breaks in the Complex I intramolecular electron transport chain.
The second useful piece of information from EPR is the intensity of the [3Fe4S]+ signal due to aconitase, that reports on the instantaneous oxidative stress burden due to ROS production as a result of MD. Upon reaction with O2•–, a non-covalently-bound iron, Fea, is lost from the active EPR-silent [4Fe4S]2+ cluster and the resulting catalytically inactive but EPR-active [3Fe4S]+ cluster exhibits a sharp and distinct, almost-isotropic signal centered at g = 2.018[90,117-120]. The magnitude of this signal has been observed to increase dramatically in tumor tissue, where excess ROS has been confirmed by other techniques, whereas in a mouse MD model the increase was moderate (125% of control)[115,79].
Chronic exposure to ROS elicits an additional response. Catalase expression is known to protect against oxidative stress, and catalase overexpression protects against ROS-mediated cellular damage[121-124]. Because catalase is not constitutively expressed at EPR-detectable levels in most tissues (liver is an exception)[79], the appearance of catalase ferriheme EPR signals at geff = 6.2 and 5.7, that flank the signal at geff = 6, provides an oxidative biomarker for sustained exposure to ROS. This signal may not be of use for monitoring MD progression, as it is for characterizing tumor growth, but could be a useful tool for evaluating therapy.
Where EPR can be much more powerful than when applied as a stand-alone tool for the characterization of MD and in subsequent therapy evaluation is when it is integrated into a comprehensive multi-technique protocol. The first and most comprehensive study of this type was the investigation of the underlying mechanism of mitochondrial dysfunction in two deoxyguanosine kinase (DGUOK; EC 2.7.1.113)-deficient rat models (“M1” and “M2”) of a genetic mitochondrial DNA depletion syndrome[79]. Mitochondrial DNA assays, histology, protein immunoblot (western blot) assays, and electron transport chain activity assays were carried out in addition to EPR measurements. Both rat models were characterized by a 90% depletion of hepatic mtDNA and a 60%-80% depletion of splenetic mtDNA, similar to those observed in the human condition for which the model was developed[125]. One of the models, M2, was ~50% deficient in brain mtDNA while the other, M1, was ~50% deficient in mtDNA in the quadriceps muscle overall although the deficiency appears not to be uniformly distributed throughout the muscle. Histological staining revealed numerous fibers in DGUOK-deficient rat muscle that were negative for Complexes II and IV whereas no such fibers were evident in wild-type (w/t) rat muscle. MRC protein expression assays indicated that Complexes II, IV, and V were expressed at w/t levels whereas Complexes I and III were expressed at about 50% of w/t. Activity assays of the MRC complexes, though, differed considerably from their expression levels. Complex II activities in both liver and muscle of DGUOK-deficient rats were close to w/t levels, as was that of Complex IV in muscle. Complexes I and III, however, exhibited very low activities, 10%-20% of w/t, lower than expected from protein expression assays. Interestingly, the activity of Complex IV in liver was < 20% of w/t despite being expressed at w/t levels. The EPR results were, therefore, considered in light of these observations.
EPR of liver indicated that the signals due to reduced [2Fe2S]+ and [4Fe4S]+ clusters of Complex I were diminished by about 50% in DGUOK-deficient rats, regardless of the cluster midpoint potential. That the same ratio of intensities of signals was observed from each of N1b, N2, N3, and N4 in w/t and MD rats indicates that the redox potential experienced by Complex I was not affected, and that the signal diminution in MD rat liver is therefore due to the proportionally diminished expression level. This result also indicates that the lower-than-expected activity of Complex I is not due to globally misfolded protein or an inability to incorporate at least the four EPR-characterized low-potential iron-sulfur clusters, but likely involves the mitochondrially-encoded ND1 subunit. The other potentially interesting results of EPR of liver were a 25% increase in the aconitase signal, and a doubling of a signal due to Mn2+. The aconitase signal suggests ongoing oxidative stress, perhaps related to the presence of the fully-reduced but partially inactive Complex I. The expression of signals due to Mn2+ has been associated with oxidative stress adaptation in bacteria[126], but any role in humans is undefined. The only clear signals associated to either of Complexes III or IV were the reduced Rieske [2Fe2S]+ cluster signal of Complex III, and the Complex IV heme a signal at geff = 3. The Rieske cluster signal from MD rats exhibited the same intensity as w/t despite lower expression and even lower activity. This observation is rationalized by the observation that Complex II is fully expressed and fully functional in MD rat liver and that electron redistribution to Complex I is thermodynamically unfavorable due to reduction of the latter. Therefore, Complex II is likely to be feeding reducing equivalents into a depleted pool of dysfunctional Complex III, ensuring complete reduction of the Rieske cluster and leading to a 50% depletion of the ferriheme a signal. The conclusion was the inability of fully-reduced Complex I to donate its electrons into the MRC may render it the source of ROS, while a fully functional Complex II is sufficient to support reduction of Complex III which, due to the low activities of Complexes III and IV themselves, is not readily reoxidized. In liver from rat models of this MD, then, both low ATP production and ROS production are likely and both sequelae would need to be addressed in a patient.
EPR of muscle paints a different picture. The human disease being modeled is characterized by weak muscle performance[125]. In rats, expression of this phenotype is far milder, possibly due to underlying metabolic corrections[79]. The reduced iron-sulfur cluster signals from each of Complex I, II, and III in MD rat muscle were diminished by 50% to 75%, with the Complex I signals depleted most. In addition, the intensity of the Complex II S3 oxidized [3Fe4S]+ cluster signal increased by a factor of three in MD rats. These data, in contrast to liver, suggest a global increase in redox potential (i.e., decrease in ATP-synthesizing thermodynamic driving force) in the muscle of DGUOK-depleted rats. A number of scenarios that might explain the phenotype and mitochondrial pathology were considered and rejected and, ultimately, a depletion of succinate combined with the inability of Complex I to release electrons from the mitochondrially-encoded ND1 subunit was considered the most likely explanation.
One perhaps surprising result was that the EPR of heart muscle in both MD and w/t rats was identical and indicated a fully reduced MRC and an absence of markers for oxidative stress. A reason for the observation may simply be that in any other case the rat would not be alive for study. The cause may be that the heart has large redundancy in the number of mitochondria in order to fully benefit from reducing equivalents from primary metabolism even when under stress.
Conclusion
EPR of biopsy tissue of a subject with suspected MD can provide some limited information, largely through the ratio of intensities of the Complex I iron-sulfur centers reporting on the ability to maintain a low redox potential, and through the observation of biomarkers for historical and ongoing ROS-mediated oxidative stress. When the EPR of such tissues can be compared with good controls, or with a yet-to-be established database of control spectra, quantitative information on the intensities of multiple MRC components can be obtained that can inform on disease mechanism. However, it is when EPR is combined with multiple complementary techniques that it becomes most useful. The primary advantages of EPR are that the sample requires no processing or fixing, and that it is relatively straightforward to determine whether the main outcome of mitochondrial dysfunction is likely to be diminished ATP synthesis, elevated ROS production, or both. One lesson from the study discussed above is that important information on an MD that is characterized in humans by poor muscle performance may be obtained from study of other tissue, e.g., liver.
Declarations
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Buchaklian AH, Helbling D, Ware SM, Dimmock DP. Recessive deoxyguanosine kinase deficiency causes juvenile onset mitochondrial myopathy. Mol Genet Metab 2012;107:92-4.
2. Dimmock DP, Zhang Q, Dionisi-Vici C, Carrozzo R, Shieh J, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat 2008;29:330-1.
3. Scharfe C, Lu HH, Neuenburg JK, Allen EA, Li GC, et al. Mapping gene associations in human mitochondria using clinical disease phenotypes. PLoS Comput Biol 2009;5:e1000374.
5. Ricci E, Moraes CT, Servidei S, Tonali P, Bonilla E, et al. Disorders associated with depletion of mitochondrial DNA. Brain Pathol 1992;2:141-7.
7. Gropman AL. Diagnosis and treatment of childhood mitochondrial diseases. Curr Neurol Neurosci Rep 2001;1:185-94.
8. Craig AK, de Menezes MS, Saneto RP. Dravet syndrome: patients with co-morbid SCN1A gene mutations and mitochondrial electron transport chain defects. Seizure 2012;21:17-20.
9. DiMauro S, Lombes A, Nakase H, Mita S, Fabrizi GM, et al. Cytochrome c oxidase deficiency. Pediatr Res 1990;28:536-41.
10. Figarella-Branger D, Pellissier JF, Scheiner C, Wernert F, Desnuelle C. Defects of the mitochondrial respiratory chain complexes in three pediatric cases with hypotonia and cardiac involvement. J Neurol Sci 1992;108:105-13.
11. Hadzsiev K, Maasz A, Kisfali P, Kalman E, Gomori E, et al. Mitochondrial DNA 11777C>A mutation associated Leigh syndrome: case report with a review of the previously described pedigrees. Neuromolecular Med 2010;12:277-84.
12. Khurana DS, Salganicoff L, Melvin JJ, Hobdell EF, Valencia I, et al. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Neuropediatrics 2008;39:8-13.
13. Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, et al. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology 1999;52:1255-64.
14. Procaccio V, Wallace DC. Late-onset Leigh syndrome in a patient with mitochondrial complex I NDUFS8 mutations. Neurology 2004;62:1899-901.
15. Wojtovich AP, Smith CO, Haynes CM, Nehrke KW, Brookes PS. Physiological consequences of complex II inhibition for aging, disease, and the mKATP channel. Biochim Biophys Acta 2013;1827:598-611.
16. Bleier L, Drose S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim Biophys Acta 2013;1827:1320-31.
17. Marí M, Morales A, Colell A, García-Ruiz C, Kaplowitz N, et al. Mitochondrial glutathione: features, regulation and role in disease. Biochim Biophys Acta 2013;1830:3317-28.
18. Carelli V, La Morgia C, Sadun AA. Mitochondrial dysfunction in optic neuropathies: animal models and therapeutic options. Curr Opin Neurol 2013;26:52-8.
19. Bindoff LA, Desnuelle C, Birch-Machin MA, Pellissier JF, Serratrice G, et al. Multiple defects of the mitochondrial respiratory chain in a mitochondrial encephalopathy (MERRF): a clinical, biochemical and molecular study. J Neurol Sci 1991;102:17-24.
20. Borchert A, Wolf NI, Wilichowski E. Current concepts of mitochondrial disorders in childhood. Semin Pediatr Neurol 2002;9:151-9.
21. Chitkara DK, Nurko S, Shoffner JM, Buie T, Flores A. Abnormalities in gastrointestinal motility are associated with diseases of oxidative phosphorylation in children. Am J Gastroenterol 2003;98:871-7.
22. Goodfellow JA, Dani K, Stewart W, Santosh C, McLean J, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes: an important cause of stroke in young people. Postgrad Med J 2012;88:326-34.
23. Gordon N. Alpers syndrome: progressive neuronal degeneration of children with liver disease. Dev Med Child Neurol 2006;48:1001-3.
25. Katzberg H, Karamchandani J, So YT, Vogel H, Wang CH. End-stage cardiac disease as an initial presentation of systemic myopathies: case series and literature review. J Child Neurol 2010;25:1382-8.
27. McDonald DG, McMenamin JB, Farrell MA, Droogan O, Green AJ. Familial childhood onset neuropathy and cirrhosis with the 4977bp mitochondrial DNA deletion. Am J Med Genet 2002;111:191-4.
28. Menezes MP, Ouvrier RA. Peripheral neuropathy associated with mitochondrial disease in children. Dev Med Child Neurol 2012;54:407-14.
31. Sakushima K, Tsuji-Akimoto S, Niino M, Saitoh S, Yabe I, et al. Adult Leigh disease without failure to thrive. Neurologist 2011;17:222-7.
32. Scaglia F. The role of mitochondrial dysfunction in psychiatric disease. Dev Disabil Res Rev 2010;16:136-43.
34. van Ekeren GJ, Stadhouders AM, Smeitink JA, Sengers RC. A retrospective study of patients with the hereditary syndrome of congenital cataract, mitochondrial myopathy of heart and skeletal muscle and lactic acidosis. Eur J Pediatr 1993;152:255-9.
35. Wallace DC, Shoffner JM, Trounce I, Brown MD, Ballinger SW, et al. Mitochondrial DNA mutations in human degenerative diseases and aging. Biochim Biophys Acta 1995;1271:141-51.
36. Wells GD, Noseworthy MD, Hamilton J, Tarnopolski M, Tein I. Skeletal muscle metabolic dysfunction in obesity and metabolic syndrome. Can J Neurol Sci 2008;35:31-40.
37. Fodale V, La Monaca E. Propofol infusion syndrome: an overview of a perplexing disease. Drug Saf 2008;31:293-303.
38. Footitt EJ, Sinha MD, Raiman JA, Dhawan A, Moganasundram S, et al. Mitochondrial disorders and general anaesthesia: a case series and review. Br J Anaesth 2008;100:436-41.
39. Gurrieri C, Kivela JE, Bojanić K, Gavrilova RH, Flick RP, et al. Anesthetic considerations in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome: a case series. Can J Anaesth 2011;58:751-63.
40. Papaioannou V, Dragoumanis C, Theodorou V, Pneumatikos I. The propofol infusion ‘syndrome’ in intensive care unit: from pathophysiology to prophylaxis and treatment. Acta Anaesthesiol Belg 2008;59:79-86.
41. Driessen J, Willems S, Dercksen S, Giele J, van der Staak F, et al. Anesthesia-related morbidity and mortality after surgery for muscle biopsy in children with mitochondrial defects. Paediatr Anaesth 2007;17:16-21.
42. Abramovich CM, Prayson RA, McMahon JT, Cohen BH. Ultrastructural examination of the axillary skin biopsy in the diagnosis of metabolic diseases. Hum Pathol 2001;32:649-55.
43. Chow CW, Thorburn DR. Morphological correlates of mitochondrial dysfunction in children. Hum Reprod 2000;15:68-78.
44. Edwards RH, Round JM, Jones DA. Needle biopsy of skeletal muscle: a review of 10 years experience. Muscle Nerve 1983;6:676-83.
45. Friedman SD, Shaw DW, Ishak G, Gropman AL, Saneto RP. The use of neuroimaging in the diagnosis of mitochondrial disease. Dev Disabil Res Rev 2010;16:129-35.
47. Gulati S, Shah T, Menon S, Jayasundar R, Kalra V. Magnetic resonance spectroscopy in pediatric neurology. Indian J Pediatr 2003;70:317-25.
48. Kyriacou K, Kyriakides T. Mitochondrial encephalomyopathies: a review of routine morphological diagnostic methods with emphasis on the role of electron microscopy. J Submicrosc Cytol Pathol 2006;38:201-8.
49. McCormick E, Place E, Falk MJ. Molecular genetic testing for mitochondrial disease: from one generation to the next. Neurotherapeutics 2013;10:251-61.
50. Micaglio G, Ceccato MB, Trevisan C, Angelini C. Quantitative histopathology in congenital myopathies. Riv Neurol 1987;57:261-8.
51. Mohri I, Taniike M, Fujimura H, Matsuoka T, Inui K, et al. A case of Kearns-Sayre syndrome showing a constant proportion of deleted mitochondrial DNA in blood cells during 6 years of follow-up. J Neurol Sci 1998;158:106-9.
52. Scaglia F, Towbin JA, Craigen WJ, Belmont JW, Smith EO, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004;114:925-31.
53. Suomalainen A. Biomarkers for mitochondrial respiratory chain disorders. J Inherit Metab Dis 2011;34:277-82.
54. Tatke M. Mitochondrial myopathies-clinicopathological features and diagnostic modalities. Indian J Pathol Microbiol 2007;50:467-77.
55. Vallance H. Biochemical approach to the investigation of pediatric mitochondrial disease. Pediatr Dev Pathol 2004;7:633-6.
56. Berardo A, Dimauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep 2010;10:118-26.
57. Darras BT, Friedman NR. Metabolic myopathies: a clinical approach; part I. Pediatr Neurol 2000;22:87-97.
58. Meunier B, Fisher N, Ransac S, Mazat JP, Brasseur G. Respiratory complex III dysfunction in humans and the use of yeast as a model organism to study mitochondrial myopathy and associated diseases. Biochim Biophys Acta 2013;1827:1346-61.
59. Boot RG, Verhoek M, de Fost M, Hollak CE, Maas M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood 2004;103:33-9.
60. Gloerich J, Wevers RA, Smeitink JA, van Engelen BG, van den Heuvel LP. Proteomics approaches to study genetic and metabolic disorders. J Proteome Res 2007;6:506-12.
61. Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkilä S, Wenz T, et al. Mitochondrial myopathy induces a starvation-like response. Hum Mol Genet 2010;19:3948-58.
62. Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A 2005;102:17687-92.
63. Kochelaev BI, Yablokov YV. The beginning of paramagnetic resonance. Singapore: World Scientific Press; 1995.
64. Weil JA, Bolton JR. Electron paramagnetic resonance: elementary theory and practical applications. 2nd ed. New York: Wiley-Interscience; 2007.
65. Hanson G. Biological magnetic resonance 28. High resolution EPR: applications to metalloenzymes and metals in medicine. New York: Springer; 2009.
66. Hanson G. Biological magnetic resonance 29. Metals in biology:application of high resolution EPR to metalloenzymes. New York: Springer; 2010.
67. Misra SK. Multifrequency electron paramagnetic resonance: theory and applications. Weinheim (Germany): Wiley VCH; 2011.
68. Misra SK. Multifrequency electron paramagnetic resonance: data and techniques. Weinheim (Germany): Wiley VCH; 2014.
69. Bennett B. EPR of cobalt-substituted zinc enzymes. Biological Magnetic Resonance 2010;29:345-70.
70. Antholine WE, Bennett B, Hanson GR. Multifrequency EPR of Cu(II). In: Misra SK, editor. Multifrequency electron paramagnetic resonance: theory and applications. Weinheim (Germany): Wiley VCH; 2011. pp. 647-718.
71. Kowalski JM, Bennett B. Spin hamiltonian parameters for Cu(II)-prion pepetide complexes from L-band electron paramagnetic resonance spectroscopy. J Am Chem Soc 2011;133:1814-23.
72. Bennett B, Hill BC. Avoiding premature oxidation during the binding of Cu(II) to a dithiolate site in BsSCO. A rapid freeze-quench EPR study. FEBS Lett 2011;515:861-4.
73. Pharr CR, Kopff LA, Bennett B, Reid SA, McMahon RJ. Photochemistry of furyl- and thienyldiazomethanes: spectroscopic characterization of triplet 3-thienylcarbene. J Am Chem Soc 2012;134:6443-54.
74. Stein N, Gumataotao N, Hajnas N, Wu R, Lankathilaka KPW, et al. multiple states of nitrile hydratase from rhodococcus equi TG328-2: structural and mechanistic insights from electron paramagnetic resonance and density functional theory studies. Biochemistry 2017;56:3066-77.
76. Neese F. Software update: the ORCA program system, version 4.0. WIREs Comput Mol Sci 2018;8:e1327.
77. Poole CP. Electron spin resonance: a comprehensive treatise on experimental techniques. 2nd ed. Mineola NY: Dover; 1983.
78. Misra SK. Relaxation of paramagnetic spins. In: Misra SK, editor. Multifrequency electron paramagnetic resonance: theory and applications. Weinheim (Germany): Wiley VCH; 2011. pp. 455-96.
79. Bennett B, Helbling D, Meng H, Jarzembowski J, Geurts AM, et al. Potentially diagnostic electron paramagnetic resonance spectra elucidate the underlying mechanism of mitochondrial dysfunction in the deoxyguanosine kinase deficient rat model of a genetic mitochondrial DNA depletion syndrome. Free Radical Biol Med 2016;92:141-51.
80. Beinert H. EPR spectroscopy of components of the mitochondrial electron-transfer system. Methods Enzymol 1978;54:133-50.
81. Haddy A, Smith G. Transition metal and organic radical components of carp liver tissue observed by electron paramagnetic resonance spectroscopy. Comparative Biochem Physiol B Comparative Biochem 1999;123:407-15.
82. Kalyanaraman B, Cheng G, Zielonka J, Bennett B. Low-temperature EPR spectroscopy as a probe-free technique for monitoring oxidants formed in tumor cells and tissues: implications in drug resistance and OXPHOS-targeted therapies. Cell Biochem Biophys 2019;77:89-98.
83. Roessler MM, King MS, Robinson AJ, Armstrong FA, Harmer J, et al. Direct assignment of EPR spectra to structurally defined iron-sulfur clusters in complex I by double electron-electron resonance. Version 2. Proc Natl Acad Sci U S A 2010;107:1930-5.
84. Medvedev ES, Couch VA, Stuchebrukhov AA. Determination of the intrinsic redox potentials of FeS centers of respiratory complex I from experimental titration curves. Biochim Biophys Acta 2010;1797:1665-71.
85. Ohnishi T, Nakamaru-Ogiso E. Were there any “misassignments” among iron-sulfur clusters N4, N5 and N6b in NADH-quinone oxidoreductase (complex I)? Biochim Biophys Acta 2008;1777:703-10.
86. Yakovlev G, Reda T, Hirst J. Reevaluating the relationship between EPR spectra and enzyme structure for the iron sulfur clusters in NADH:quinone oxidoreductase. Proc Natl Acad Sci U S A 2007;104:12720-5.
87. Beinert H, Ackrell BA, Kearney EB, Singer TP. Iron-sulfur components of succinate dehydrogenase: stoichiometry and kinetic behavior in activated preparations. Eur J Biochem 1975;54:185-94.
88. Maguire JJ, Johnson MK, Morningstar JE, Ackrell BA, Kearney EB. Electron paramagnetic resonance studies of mammalian succinate dehydrogenase. Detection of the tetranuclear cluster S2. J Biol Chem 1985;260:10909-12.
89. Johnson MK, Morningstar JE, Bennett DE, Ackrell BA, Kearney EB. Magnetic circular dichroism studies of succinate dehydrogenase. Evidence for [2Fe-2S], [3Fe-xS], and [4Fe-4S] centers in reconstitutively active enzyme. J Biol Chem 1985;260:7368-78.
90. Kennedy MC, Antholine WE, Beinert H. An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J Biol Chem 1997;272:20340-7.
91. Salerno JC, Ohnishi T. Studies on the stabilized ubisemiquinone species in the succinate-cytochrome c reductase segment of the intact mitochondrial membrane system. Biochem J 1980;192:769-81.
92. Orme-Johnson NR, Hansen RE, Beinert H. Electron paramagnetic resonance-detectable electron acceptors in beef heart mitochondria. J Biol Chem 1974;249:1928-39.
93. Rieske JS, MacLennan DH, Coleman R. Isolation and properties of an iron-protein from the (reduced coenzyme Q)-cytochrome C reductase complex of the respiratory chain. Biochem Biophys Res Commun 1964;15:338-44.
94. Trumpower BL, Edwards CA. Purification of a reconstitutively active iron-sulfur protein (oxidation factor) from succinate. cytochrome c reductase complex of bovine heart mitochondria. J Biol Chem 1979;254:8697-706.
95. Siedow JN, Power S, de la Rosa FF, Palmer G. The preparation and characterization of highly purified, enzymically active complex III from baker’s yeast. J Biol Chem 1978;253:2392-9.
96. Aasa R, Albracht PJ, Falk KE, Lanne B, Vänngard T. EPR signals from cytochrome c oxidase. Biochim Biophys Acta 1976;422:260-72.
97. Kroneck PM, Antholine WE, Kastrau DH, Buse G, Steffens GC, et al. Multifrequency EPR evidence for a bimetallic center at the CuA site in cytochrome c oxidase. FEBS Lett 1990;268:274-6.
98. Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys J 2009;96:1388-98.
99. Hagen WR. EPR of non-Kramers doublets in biological systems: characterization of an S = 2 system in oxidized cytochrome c oxidase. Biochim Biophys Acta 1982;708:82-98.
100. Cooper CE, Salerno JC. Characterization of a novel g’ = 2.95 EPR signal from the binuclear center of mitochondrial cytochrome c oxidase. J Biol Chem 1992;267:280-5.
101. Torii K, Iizuka T, Ogura Y. Magnetic susceptibility and EPR measurements of catalase and its derivatives. A thermal equilibrium between the high- and low-spin states in the catalase-azide compound. J Biochem 1970;68:837-41.
102. Bomba M, Camagna A, Cannistraro S, Indovina PL, Samoggia P. EPR study of serum ceruloplasmin and iron transferrin in myocardial infarction. Physiol Chem Phys 1977;9:175-80.
103. Harris DC. Different metal-binding properties of the two sites of human transferrin. Biochemistry 1977;16:560-4.
104. Dunne J, Caron A, Menu P, Alayash AI, Buehler PW, et al. Ascorbate removes key precursors to oxidative damage by cell-free haemoglobin in vitro and in vivo. Biochem J 2006;399:513-24.
105. Kumar P, Bulk M, Webb A, van der Weerd L, Oosterkamp TH, et al. A novel approach to quantify different iron forms in ex-vivo human brain tissue. Sci Rep 2016;6:38916.
106. Langley M, Ghosh A, Charli A, Sarkar S, Ay M, et al. Mito-apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in mitopark transgenic mice. Antioxid Redox Signal 2017;27:1048-66.
107. Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, et al. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Rad Biol Med 2010;49:1674-84.
108. Cheng G, Zielonka M, Dranka B, Kumar SN, Myers CR, et al. Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: Potentials, pitfalls, and the future. J Biol Chem 2018;293:10363-80.
109. Kalyanaraman B, Cheng G, Hardy M, Ouari O, Bennett B, et al. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol 2018;15:347-62.
110. Siebers EM, Choi MJ, Tinklenberg JA, Beatka MJ, Ayres S, et al. Sdha+/- rats display minimal muscle pathology without significant behavioral or biochemical abnormalities. J Neuropathol Exp Neurol 2018;77:665-72.
111. Sethumadhavan S, Whitsett J, Bennett B, Ionova IA, Pieper GM, et al. Increasing tetrahydrobiopterin in cardiomyocytes adversely affects cardiac redox state and mitochondrial function independently of changes in NO production. Free Radic Biol Med 2016;93:1-11.
112. Levenberg K. A method for the solution of certain non-linear problems in least squares. Quarterly Applied Mathematics 1944;2:164-8.
113. Marquardt D. An algorithm for least-squares estimation of nonlinear parameters. SIAM J Applied Mathematics 1963;11:431-41.
114. Kanzow C, Yamashita N, Fukushima M. Levenberg-Marquardt methods with strong local convergence properties for solving nonlinear equations with convex constraints. J Comput Appl Math 2004;172:375-97.
115. Cheng G, Pan J, Podsiadly R, Zielonka J, Garces AM, et al. Increased formation of reactive oxygen species during tumor growth: Ex vivo low-temperature EPR and in vivo bioluminescence analyses. Free Radic Biol Med 2020;147:167-74.
116. Unden G, Bongaerts J. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim Biophys Acta 1997;1320:217-34.
117. Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem 1994;269:29405-8.
118. Vasquez-Vivar J, Kalyanaraman B, Kennedy MC. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J Biol Chem 2000;275:14064-9.
119. Tortora V, Quijano C, Freeman B, Radi R, Castro L. Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radical Biol Med 2007;42:1075-88.
120. Bulteau AL, Ikeda-Saito M, Szweda LI. Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry 2003;42:14846-55.
121. Selvaratnam J, Robaire B. Overexpression of catalase in mice reduces age-related oxidative stress and maintains sperm production. Exp Gerontol 2016;84:12-20.
122. Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005;308:1909-11.
123. Meilhac O, Zhou M, Santanam N, Parthasarathy S. Lipid peroxides induce expression of catalase in cultured vascular cells. J Lipid Res 2000;41:1205-13.
124. Bai J, Rodriguez AM, Melendez JA, Cederbaum AI. Overexpression of catalase in cytosolic or mitochondrial compartment protects HepG2 cells against oxidative injury. J Biol Chem 1999;274:26217-24.
125. Dimmock D, Tang LY, Schmitt ES, Wong LJ. Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin Chem 2010;56:1119-27.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Bennett B. Cryogenic electron paramagnetic resonance spectroscopy of flash-frozen tissue for characterization of mitochondrial disease. J Transl Genet Genom 2020;4:36-49. http://dx.doi.org/10.20517/jtgg.2020.17
AMA Style
Bennett B. Cryogenic electron paramagnetic resonance spectroscopy of flash-frozen tissue for characterization of mitochondrial disease. Journal of Translational Genetics and Genomics. 2020; 4(2): 36-49. http://dx.doi.org/10.20517/jtgg.2020.17
Chicago/Turabian Style
Bennett, Brian. 2020. "Cryogenic electron paramagnetic resonance spectroscopy of flash-frozen tissue for characterization of mitochondrial disease" Journal of Translational Genetics and Genomics. 4, no.2: 36-49. http://dx.doi.org/10.20517/jtgg.2020.17
ACS Style
Bennett, B. Cryogenic electron paramagnetic resonance spectroscopy of flash-frozen tissue for characterization of mitochondrial disease. J. Transl. Genet. Genom. 2020, 4, 36-49. http://dx.doi.org/10.20517/jtgg.2020.17
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 21
likes
Like This Article 21
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.