
Spectrum of MECP2 mutations in Indian females with Rett Syndrome - a large cohort study

Abstract
Aim: This study aimed to characterize MECP2 gene variants in Indian female patients with classical Rett syndrome (RTT).
Methods: Seventy-two patients fulfilling the revised diagnostic criteria of classical RTT were enrolled and exons 2-4 of MECP2 gene were analyzed by Sanger sequencing followed by quantitative analysis using MLPA. Bioinformatic analysis was done using different software packages to predict the effect of sequence variations on the function of the MeCP2 protein.
Results: A heterogeneous spectrum of MECP2 sequence variants including 13 novel variants was identified with a detection rate of 98.6%. The majority of the variants were distributed in the functional domain of MECP2 with most missense variants clustered in methyl binding domain and truncating variants in interdomain and transcription repression domain of MECP2. Genotype-phenotype correlations revealed that patients carrying early truncating variants presented with a more severe phenotype.
Conclusion: RTT is a childhood neurodevelopmental disorder primarily affecting females. It is caused by mutations in the Methyl-CpG-Binding Protein 2 gene (MECP2 ), an important regulator of gene expression, located at Xq28. Variants in MECP2 can be identified in 95%-97% of individuals with Classical RTT using a combination of molecular techniques. This large cohort study from India showed the highest detection rate of MECP2 variants in classical RTT patients, emphasizing the importance of using diagnostic criteria and having a multidisciplinary team in the assessment of RTT patients, which can further help provide diagnostic testing, genetic counseling, and prenatal testing.
Keywords
Introduction
Rett syndrome (RTT, OMIM #312750) is a childhood neurodevelopmental disorder primarily affecting females. It is caused by mutations in the Methyl-CpG-Binding Protein 2 gene (MECP2, OMIM *300005), an important regulator of gene expression, located at Xq28[1].
Developmental regression is a hallmark of RTT, the ongoing pathology of which is still being unraveled. Symptoms include loss of acquired skills, especially in relation to communicative and motor performance. Clinical developmental profiles, non-specific early in life, become more specific later. To support clinical diagnosis, a staging system has been developed as a framework that delineates the evolving symptoms. This includes stages of early-onset stagnation, rapid developmental regression, a pseudo-stationary stage, and late motor deterioration. We do not yet fully understand the biological pathways underlying the outward presentations of the RTT[2]. The multi-functionality of MECP2 suggests there are many downstream pathways that are interesting for understanding the pathophysiology of RTT.
Variants in MECP2 can be identified in 95%-97% of individuals with Classical RTT, using a combination of mutation detection techniques[3]. Classical RTT is characterized by apparently normal early development, arrest of developmental progress at 6-18 months followed by regression of social contact, language, and hand skills. However, thereafter, improvements in social behavior and eye contact have been observed. The most recent revision of the clinical criteria for diagnosis of RTT[4] allows for a broader interpretation of regression and partial recovery than was previously acknowledged and has led to increased understanding of the disease[5]. Clinicians should be aware of these criteria, for counseling of families as they seek to understand the stages their child will encounter and for the application of management strategies that may help to ameliorate or compensate for loss of skills at the different stages across the lifespan. A review of the literature of mutation analysis in large cohorts of RTT patients in Western populations indicates that the majority are sequence variations and only a small proportion of cases have large deletions/duplications[6,7]. To the best of our knowledge, there are only two studies on mutation spectrum of RTT from India, including both typical and atypical RTT[8,9] and until now no study has been reported on a large cohort of classical RTT patients describing the spectrum of MECP2 sequence variations and to evaluate the genotype-phenotype correlations based on the mutation spectrum. The objectives of the present study were: (1) to study the clinical phenotype of Indian patients with classical RTT; (2) to identify the spectrum of MECP2 sequence variations in a large cohort of Indian RTT patients and determine genotype-phenotype correlation, if any; and (3) to predict the effects of MECP2 variations on MeCP2 Protein using bioinformatics.
Methods
Seventy-two sporadic classical RTT patients (all females) were included in this study from Pediatric OPD, Pediatric wards, Pediatric Neurology and Medical Genetics services of the All India Institute of Medical Sciences New Delhi, India. Patients were defined as classical when they showed a period of regression and fulfilled all the main inclusion criteria (partial or complete loss of acquired purposeful hand skills; partial or complete loss of acquired spoken language; gait abnormalities; impaired or absence of ability to walk; stereotypic hand movements) as per the revised diagnostic criteria for classical RTT[4]. The patients not fulfilling the major criteria and having the clinical signs mentioned in exclusion criteria[4] were excluded from the study. Exclusion criteria were: evidence of brain injury secondary to perinatal or postnatal events, neuro-metabolic disorders or infection causing neurological problems. Additionally, children having grossly abnormal psychomotor development in first six months of life were also excluded. Ethical approval for the present study was taken from the Ethics Committee of the Institute. Proper information about the study was given to all families and written informed consent was obtained from the parent/guardian. Enrolled patients represented all regions of India, with the majority from northern India. Patients were evaluated by a team comprising of a clinical geneticist, a pediatric neurologist, and a child psychologist before inclusion in the study. All clinical details were recorded in a predesigned proforma.
Five-milliliter blood samples were collected from all patients in EDTA vacutainer and DNA was extracted by standard phenol-chloroform method. DNA samples were analyzed using bidirectional Sanger sequencing for sequence variations followed by quantitative analysis using Multiplex Ligation-dependent Probe Amplification (MLPA) for deletion/duplication analysis of MECP2 gene. The gene nomenclature used was according to guidelines of HGNC (Hugo Genome Nomenclature Committee) and the recommended sequence variant nomenclature of HGVS (Human Genome Variation Society)[10]. Any change found in DNA sequence of the RTT patients was also analyzed in their family members (except three cases) to confirm its origin. The sequence variant profile was compared with the clinical presentation to generate genotype-phenotype correlations. All novel variants identified in this study were submitted to National Centre for Biotechnology Information (NCBI) GenBank (http://www.ncbi.nlm.nih.gov/GenBank) and RettBASE: IRSF MECP2 Variation Database (http://mecp2.chw.edu.au)[11].
MECP2 screening
The coding region of exons 2-4 of MECP2 gene (transcript 1, MeCP2_e2) including flanking exon/intron boundaries was amplified by PCR using seven overlapping primer sets (2.1, 2.2, 3.1, 3.2, 3.3, 4.1, and 4.2) of MECP2 gene published elsewhere[1,12]. PCR amplification was performed in a final volume of 25 µL containing 10× PCR buffer with 1.5 mM MgCl2, 0.25 mM dNTPs, 0.625 U Taq polymerase, 1 pM/µL each of forward and reverse primer, and 50 ng of DNA. All samples were analyzed by direct bidirectional Sanger sequencing. The data were interpreted and compared with reference sequence of MECP2 (NM_004992.3) gene. MLPA technique was used to screen the RTT patients who were negative for MECP2 sequence variations on DNA sequencing to check for gross rearrangements[6,13,14]. SALSA MLPA kits P015-D2 and P015-E2 (MRC-Holland, Amsterdam, The Netherlands) were used. All cases with positive or aberrant results were rerun in a second MLPA reaction for confirmation.
Bioinformatic analysis
In the present study, various bioinformatic software packages were used for prediction of pathogenicity of MECP2 sequence variations. Prediction tools such as PolyPhen (Polymorphism Phenotyping) (http://genetics.bwh.harvard.edu/pph/), SIFT (Sorting Tolerant from Intolerant) (http://sift.jcvi.org/), and SNPs3D (http://www.snps3d.org/) were used to predict if the non-synonymous particular variants are likely to be deleterious. Prediction of the effect of synonymous variants was done using Codon Usage Database (www.kazusa.or.jp/codon/). In-silico splice site analysis was done using Alamut (http://www.interactive-biosoftware.com/alamut/) for prediction of splice-site variants. We used MeCP2_e2 transcript of MeCP2 for pair wise alignment and the sequences were derived from NCBI HomoloGene (http://www.ncbi.nlm.nih.gov/homologene). The alignments were produced using ClustalW (http://www.ebi.ac.uk/Tools/clustalw/).
Statistical analysis
We used Analysis of Variance (ANOVA) to compare the group mean score on dependent variables, covarying for age. Categorical variables were analyzed for significant associations using Pearson χ2. STATA version 9 was used for all statistical analysis.
Results
Phenotypic features
All patients had apparently normal prenatal and perinatal history with normal early psychomotor development. We did not have head circumference records of all patients at birth, but all had microcephaly at the time of inclusion. The mean age of onset of symptoms in classical RTT patients was 16 ± 4.6 months (range: 6-30) and the median age was 16 months. The mean age at diagnosis of classical RTT patients was 54 ± 34.9 months (range: 18-186 months) while the median age was 42 months. All patients had partial or complete loss of acquired purposeful hand skills. Stereotypic hand movements were present in all patients and the common hand stereotypes observed in our patients were hand wringing, washing, mouthing, clenching, finger rolling, tapping, and clapping. All patients had partial or complete loss of acquired language and most patients spoke only monosyllables or babbling. All patients had gait problems, of which 40% (29/72 patients) could not walk, 39% (28/72 patients) had an impaired gait, and 21% (15/72 patients) could walk with support.
MECP2 sequence variants
Using DNA sequencing, we identified MECP2 sequence variants in coding region with a frequency of 88.9%. Using MLPA, large deletions of MECP2 gene were identified in 9.7% of patients. One patient was identified with intronic variant and no other sequence variant was identified in this patient. In total, 38 different types of MECP2 sequence variations (25 reported and 13 novel) were identified in all 72 classical RTT patients [Table 1].
List of MECP2 sequence variants identified in RTT patients in the present study
| Exon/Intron | Nt.a change | A.Aa change | Origin | Domain | Recr. |
|---|---|---|---|---|---|
| intron 2 | c.377+22C>G# | - | de-novo | - | 1 |
| 2.1 | c.50dupA | p.D17Fs | de-novo | NTR | 1 |
| 2.2 | c.210C>T# | p.S70S | de-novo | NTR | 1 |
| 2.2 | c.274G>T | p.G92X | de-novo | MBD | 1 |
| 2.2 | c.316C>T | p.R106W | de-novo | MBD | 2 |
| 2.2 | c.375C>A# | p.I125I | de-novo | MBD | 1 |
| intron 3 | c.378-74C>T# | - | de-novo | - | 2 |
| 3.1 | c.397C>T | p.R133C | de-novo | MBD | 5 |
| 3.1 | c.401C>G | p.S134C | de-novo | MBD | 1 |
| 3.1 | c.413T>C | p.L138S | de-novo | MBD | 1 |
| 3.1 | c.455C>G | p.P152R | de-novo | MBD | 3 |
| 3.1 | c.468C>G | p.D156E | de-novo | MBD | 1 |
| 3.1 | c.471 C>G | p.F157L | de-novo | MBD | 1 |
| 3.1 | c.473C>T | p.T158M | de-novo | MBD | 8 |
| 3.1 | c.484dupA | p.R162fs | de-novo | MBD | 1 |
| 3.1 | c.502C>T | p.R168X | de-novo | ID | 7 |
| 3.2 | c.683C>G# | p.T228S | de-novo | TRD | 1 |
| 3.2 | c.763C>T | p.R255X | de-novo | TRD/NLS | 5 |
| 3.2 | c.784C>T | p.Q262X | de-novo | TRD | 1 |
| 3.2 | c.806delG | p.G269fs | de-novo | TRD | 5 |
| 3.3 | c.808C>T | p.R270X | de-novo | TRD/NLS | 8 |
| 3.3 | c.808delC | p.R270fs | de-novo | TRD/NLS | 1 |
| 3.3 | c.834C>T# | p.A278A | de-novo | TRD/NLS | 1 |
| 3.3 | c.856_859delAAAG | p.K286fs | de-novo | TRD | 1 |
| 3.3 | c.869dupA | p. E290fs | de-novo | TRD | 1 |
| 3.3 | c.880 C>T | p.R294X | de-novo | TRD | 1 |
| 3.3 | c.916C>T | p.R306C | de-novo | TRD | 5 |
| 4.1 | c.1151_1188del38 | p.P384fs | de-novo | CTR | 1 |
| 4.1 | c.1157_1200del44 | p.L386Fs | de-novo | CTR | 2 |
| 4.1 | c.1180G>A# | p.E394K | de-novo | CTR | 1 |
| 4.2 | c.1284C>T# | p.G428G | de-novo | CTR | 1 |
| 4.2 | c.1452dupAG | p.V485fs | de-novo | CTR | 1 |
| 4.2 | c.1455dupTA | p. S486fs | de-novo | CTR | 1 |
| ex 3.1 upto ex 4.1 | c.378-?_c.1017+?del* | - | de-novo | MBD, TRD, CTR | 2 |
| int 2 upto 3’UTR | c.27-?_c.1396+?del* | - | de-novo | MBD, TRD, CTR | 2 |
| ex 4.1 upto 3’UTR | c.1396-?_c.X5349+?del*; | - | N.T | MBD, TRD, CTR | 1 |
| int 2 upto ex 4.1 | c.27-?_c.1017+?del* | - | N.T | MBD, TRD, CTR | 1 |
| 3’UTR | c.X5349-?_c.5349+?del* | - | N.T | 3’UTR | 1 |
Six recurrent sequence variants (p.P152R, p.T158M, p.R168X, p.R270X, p.G269fs, and p.R306C) were identified in 36/72 RTT patients. All of these variants were identified in heterozygous form. The p.T158M and p.R270X were the most recurrent sequence variants observed, followed by p.R168X, p.G269fs, p.R306C, and p.P152R variants [Table 2]. Twenty-nine patients (40.3%) were found positive with other uncommon heterozygous MECP2 sequence variants and one patient (1.38%) was identified with only one intronic variant c.378-74C>T [Table 1].
Common MECP2 sequence variants found in classical RTT patients in present study
| Exon | Domain | Type | Nt.a change | A.Aa change | Recurrence |
|---|---|---|---|---|---|
| 3.1 | MBD | MS | c.455C>G | p.P152R | 3 |
| 3.1 | MBD | MS | c.473C>T | p.T158M | 8 |
| 3.1 | ID | NS | c.502C>T | p.R168X | 7 |
| 3.3 | TRD-NLS | FS | c.806delG | p.G269fs | 5 |
| 3.3 | TRD-NLS | NS | c.808C>T | p.R270X | 8 |
| 3.3 | TRD | MS | c.916C>T | p.R306C | 5 |
Seven patients (six negative for MECP2 sequence variants on DNA sequencing and one patient who was identified with only one intronic variant c.378-74C>T) were screened using MLPA analysis for ruling out large deletion/duplications of MECP2 gene that cannot be identified using Sanger sequencing. In six out of seven (8.33%) RTT patients, large deletions of one or more contiguous exons, especially of exon 3 and 4 of MECP2, gene were identified. In one patient, an undefined deletion in 3’UTR of MECP2 gene was detected [Table 1], and it was confirmed that there was no sequence change at the probe hybridization site, thus it most likely was a real deletion.
Most of the mutations or sequence variants identified in patients were de novo and were not found in the family members except two cases where different mutation or variant was identified in family members. In one family, the asymptomatic mother of one RTT with p.D17fs mutation was found carrying missense mutation p.H51Q (GenBank accession No. GU812286.1) of MECP2. In another family, the patient was carrying p.R106W mutation, whereas her asymptomatic father was a carrier of intronic c.378-74C>T variant of MECP2 gene. However, in three cases, the origin of novel large deletions could not be confirmed as the parents were not available for testing.
Thirteen novel MECP2 variants were identified in this study in the RTT patients [Table 3], which included four large deletions, six frameshift [p.D17fs (GenBank accession No. FJ212168.1), p.R162fs (GenBank accession No. GU812285.1), p.E290fs (GenBank accession No. GQ203293.1), p.P384fs (GenBank accession No. FJ212171.1), p.V485fs (GenBank accession No. HQ141378), and p.S486fs (GenBank accession No. GQ203295.1)], one missense p.L138S, one synonymous p.G428G (GenBank accession No. FJ212169.1), and one nonsense (p.G92X) variant [Table 3 and Figure 1].
List of novel MECP2 sequence variants identified in RTT patients in present study
| Exon/Intron | Nt.a change | A.Aa change | Origin | Domain | Recr. |
|---|---|---|---|---|---|
| 2.1 | c.50dupA# | p.D17Fs | de-novo | NTR | 1 |
| 2.2 | c.274G>T | p.G92X | de-novo | MBD | 1 |
| 3.1 | c.413T>C | p.L138S | de-novo | MBD | 1 |
| 3.1 | c.484dupA | p.R162fs | de-novo | MBD | 1 |
| 3.3 | c.869dupA | p. E290fs | de-novo | TRD | 1 |
| 4.1 | c.1151_1188del38# | p.P384fs | de-novo | CTR | 1 |
| 4.2 | c.1284C>T# | p.G428G | de-novo | CTR | 1 |
| 4.2 | c.1452dupAG | p.V485fs | de-novo | CTR | 1 |
| 4.2 | c.1455dupTA | p. S486fs | de-novo | CTR | 1 |
| ex 3.1 upto ex 4.1 | c.378-?_c.1017+?del* | - | de-novo | MBD, TRD, CTR | 2 |
| ex 4.1 upto 3’UTR | c.1396-?_c.X5349+?del* | - | N.T | MBD, TRD, CTR | 1 |
| int 2 upto ex 4.1 | c.27-?_c.1017+?del* | - | N.T | MBD, TRD, CTR | 1 |
| 3’UTR | c.X5349-?_c.5349+?del* | - | N.T | 3’UTR | 1 |
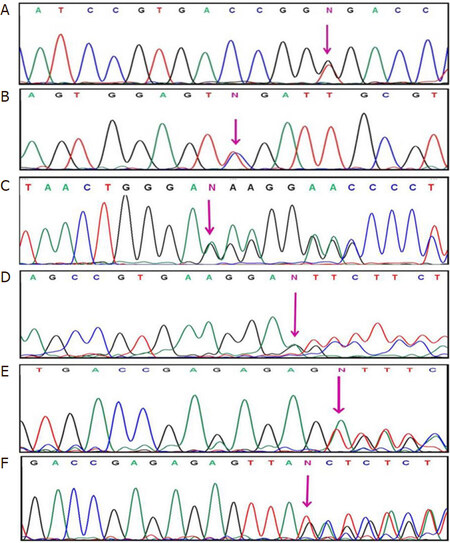
Figure 1. Sequencing chromatograms of novel MECP2 variants identified in RTT patients in present study: A: frameshift variant p.G92X; B: missense variant p.L138S; C: frameshift variant p.R162fs; D: frameshift variant p. E290fs; E: frameshift variant p.V485fs; and F: frameshift variant p. S486fs
Seven RTT patients were found carrying more than one variant in MECP2 gene [Table 1]. One of the RTT patients carrying p.L386fs mutation was also carrying a novel synonymous change p.G428G (c.1284C>T). In two patients carrying mutation p.R270X, one of them was also carrying synonymous change p.S70S (c.210C>T) and the other was carrying an intronic variation c.377+22C>G [Table 1]. Another RTT patient with p.R306C (c.916C>T) mutation was also carrying an intronic variation c.378-74C>T in intron 3 of MECP2 gene [Table 1]. Another patient with mutation p.P152R was found to be carrying another missense change p.T228S in MECP2 gene. One of the patients with p.R255X mutation was found to be carrying another synonymous change p.I125I and another patient carrying p.K286fs mutation was carrying two more variants p.E394K and p.A278A.
Most of the MECP2 variants identified in present study were missense (28%), followed by frameshift (25.6%), large deletions (17.9%), nonsense (15.3%), synonymous (10.2%), and intronic (5.1%). The majority of MECP2 sequence variations were found in Methyl Binding Domain (MBD), followed by Transcription Repression Domain (TRD), C-terminal region (CTR), N-terminal region, and Interdomain (ID). Thus, they were distributed over all the domains of MECP2[Figure 2].
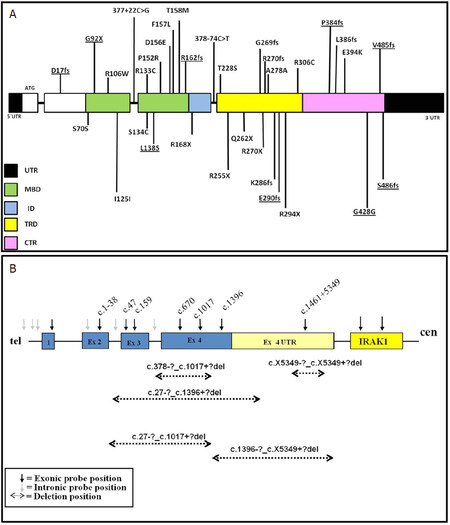
Figure 2. A: diagrammatic representation of MECP2 sequence variants identified in this study (novel variants are underlined); and B: diagrammatic representation of MECP2 deletions found in the present study
Bioinformatic analysis
The majority of missense variants in the present study were predicted as deleterious, with the exception of variants such as p.T228S that were predicted as benign or non-deleterious using prediction Polyphen, SNPs3D, and SIFT; as the patient carrying this change was carrying another deleterious change p.P152R, it can be considered a neutral change. Based on the findings of codon usage database, the synonymous variant p.I125I changed the most preferred codon ATC to a least preferred codon ATA, whereas, in the sequence variations p.S70S, p.G428G, and p.A278A, the most preferred codons were changed to less preferred codons, hence were likely predicted to be associated with disease in some way, although it is difficult to explain the implication of synonymous variants in the disease process.
The sequence of human MeCP2 was aligned with MeCP2 of cattle, dog, and mouse, and it was found that most of the missense and truncating variants identified were affecting the conserved residues of the protein, thus predicted to be affecting the protein in some way.
Genotype-phenotype correlations
Significant correlation was seen for type of sequence variation and development of clinical features. In the present study, the recurrent nonsense sequence variations p.R270X, p.R168X, and p.R255X were found significantly associated with development of scoliosis (Pearson χ2; P = 0.002) and presence of mild facial dysmorphism (Pearson χ2 test; P = 0.006). Patients with early truncating sequence variations developed poor verbal communication as compared to patients with missense sequence variations (Pearson χ2 test; P = 0.000). The ability to walk was severely deteriorated in patients with early truncating variants as compared to patients with missense variants and large deletions (Pearson χ2 test; P = 0.011). The loss of purposeful use of hands was observed to be significantly associated with missense variants compared to truncating variants (Pearson χ2 test; P = 0.032). However, generally, no significant difference was observed with the type of sequence variation and the age of onset of symptoms; exceptions were observed in patients with p.R270X and p.R168X with earlier onset of symptoms [Table 4].
Genotype-phenotype correlations of RTT patients based on the profile of common sequence variants
| Clinical features | p.T158M (n = 8) | p.R270X (n = 8) | p.R168X (n = 7) | p.R255X (n = 5) | p.R133C (n = 5) | p.R306C (n = 5) | p.G269fs (n = 5) |
|---|---|---|---|---|---|---|---|
| AD (months) Mean ± SD Median (IQR) | 37.9 ± 15 36 (21-72) | 57.8 ± 55.5 33 (18-186) | 58.3 ± 36.9 60 (18-186) | 56.4 ± 16.8 48 (42-84) | 55.2 ± 22.2 60 (30-84) | 49.2 ± 9.9 54 (36-60) | 69.8 ± 60.8 54 (19-168) |
| AO (months) Mean ± SD Median (IQR) | 16.5 ± 3.7 16 (12-24) | 13 ± 2.1 12 (12-18) | 13.9 ± 5.6 14 (12-18) | 16.4 ± 3.3 18 (12-20) | 20 ± 3.7 18 (16-24) | 16.8 ± 5.8 18 (8-24) | 16.4 ± 3.3 18 (12-20) |
| Speech problem | 8 | 8 | 7 | 5 | 5 | 5 | 5 |
| Hand stereotypes | 8 | 8 | 7 | 5 | 5 | 5 | 5 |
| Some hand/finger use | 0 | 0 | 0 | 0 | 1 | 2 | 0 |
| Microcephaly | 5 | 7 | 6 | 5 | 4 | 3 | 4 |
| Neuroregression | 6 | 6 | 6 | 5 | 5 | 5 | 5 |
| Constipation | 6 | 1 | 6 | 4 | 0 | 1 | 1 |
| Walking capability Ataxic With support No | 3 3 2 | 2 1 5 | 2 2 3 | 1 0 4 | 5 0 0 | 4 1 0 | 2 2 1 |
| Breathing problems | 3 | 4 | 2 | 3 | 2 | 3 | 2 |
| Abn imaging study | 0 | 0 | 0 | 0 | 1 | 2 | 1 |
| Abn muscle tone | 2 | 4 | 2 | 0 | 2 | 1 | 2 |
| Muscle atrophy | 3 | 4 | 2 | 3 | 0 | 1 | 1 |
| Growth retardation | 7 | 5 | 4 | 4 | 1 | 1 | 3 |
| Sleep problem | 5 | 5 | 3 | 5 | 3 | 2 | 3 |
| Abn EEG | 2 | 4 | 3 | 5 | 3 | 2 | 2 |
| Seizures | 2 | 4 | 3 | 3 | 3 | 1 | 2 |
| Scoliosis | 1 | 5 | 3 | 3 | 0 | 0 | 1 |
| Hearing impairment | 0 | 0 | 1 | 0 | 0 | 1 | 1 |
| Mild dysmorphism | 0 | 2 | 2 | 2 | 0 | 0 | 0 |
| Opthal. impairment | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| Mortality | 1 | 0 | 0 | 2 | 0 | 1 | 0 |
The mortality was also higher in RTT patients with nonsense variants than missense variants, although we were unable to find any statistical significance due to small number of patients. Most of the missense sequence variations in our study were clustered in MBD of MeCP2 and the early truncating sequence variations were similarly clustered in TRD of MeCP2 (Pearson χ2 test; P = 0.000).
Discussion
Until now, few reports on variant analysis of MECP2 gene in classical RTT have been published from India[8,9,15], thus there are scarce data regarding mutation spectrum and genotype-phenotype correlations. The present study evaluated 72 Classical RTT females based on the revised diagnostic criteria and revealed a heterogeneous spectrum of sequence variants including 13 novel variants with a detection rate of 98.6% using a combination of Sanger sequencing and MLPA, which is higher compared to other studies on mutation spectrum of RTT. The results of the present study emphasize the need for a careful and meticulous clinical evaluation that is likely to select appropriate cases with a good yield on molecular testing, which is important in resource constrained settings.
It has been reported that MECP2 variants can be detected with a frequency of more than 95%-97% in classical RTT by screening coding region and flanking intronic regions of MECP2 gene using PCR based techniques[3,16], but these methods do not detect gross rearrangements, which could be present in a significant proportion of classical RTT patients[5]. Several groups have identified gross rearrangements using quantitative analysis of MECP2 using MLPA in the patients where the cause of RTT remains unknown after sequencing[6,13,14,17].
In the present study, we were able to find MECP2 sequence variations in overall 90.3% of RTT patients using DNA sequencing. Using MLPA analysis, we were able to detect large putative deletions of MECP2 in all the classical RTT patients, which were negative on DNA sequencing. MLPA increased the detection rate of MECP2 sequence variants identified in RTT patients from 90.3% to 98.6%. We propose that MLPA analysis of MECP2 is crucial and needs to be performed in classical RTT patients. Large deletions can be missed using DNA sequencing and reaffirms the view that large MECP2 deletions are an important cause of classical RTT[6,13]. In this study, the majority of the RTT patients were carrying the C>T transitions, supporting the reported literature[12].
Data from different western studies have shown MECP2 sequence variation frequency between 70% and 97% in classical RTT[1,3,7,12,16,18]. A literature search revealed many studies on RTT from Asia that reported MECP2 sequence variation frequency of 50%-92.5% in classical RTT patients[19-23]. In the two Indian studies published thus far, the detection rates of MECP2 variations was lower as compared to our study[8,9]. The study by Lallar et al.[9], which included 19 RTT patients (14 classical and 5 atypical), reported a detection rate of 93% in classical RTT girls using a combination of Sanger sequencing followed by MLPA analysis, supporting our findings. In the other Indian study by Das et al.[8], investigating 90 individuals with suspected RTT phenotype, 19 different MECP2 mutations and polymorphisms were identified in 27/90 (30%) patients while the rest remained uncharacterized.
The high rate of MECP2 sequence variation detected in the present study compared to the data from other Asian, Indian, and western studies can be explained by the fact that a strict inclusion/exclusion of classical RTT patients was adopted based on revised clinical diagnostic criteria of RTT[4] and involvement of a multidisciplinary team for clinical evaluation of the patients. Our study supports the previous findings that clinical stringency based on diagnostic criteria can increase the mutation detection rate in RTT patients and emphasizes the importance of diagnostic criteria in the assessment of RTT patients[24].
The worldwide reported eight hotspot MECP2 sequence variants p.R106W, p.R133C, p.T158M, p.R168X, p.R255X, p.R270X, p.R294X, and p.R306C were identified with a frequency of 57% in our study, which is similar to previously reported western and Asian studies[3,7]. The hotspot variant p.R294X identified recurrently in western population was found in only one patient in the present study, whereas other hotspot variants, namely p.P152R, p.G269fs, and p.L386fs, were identified in more than one patient. In another Indian study by Das et al.[8], which included 90 patients (suspected RTT patients), showed MECP2 sequence variations in only 30% of patients (27/90 patients), of which seven hotspot variants, namely p.R106W, p.R133C, p.T158M, p.R168X, p.R255X, p.R270X, and p.R306C (sequence variant p.R294X was not reported in this study), were identified with frequency of 51.8%. The other Indian study by Lallar et al.[9] reported four common hotspot variants (p.R168X, p.T158M, p.R306C, and p.R255X). The present study indicates that the variant p.R294X may not be a hotspot sequence variation in Indian RTT patients and instead p.G269fs, p.P152R, and p.L386fs, may be the hotspot sequence variations along with the seven other recurrent variants identified. Multiple recurrences of these three variant (p.G269fs, p.P152R, and p.L386fs) in the present study indicate that these variants could be the hotspot variants specific to Indian population. Larger studies from India are needed to confirm and support these findings.
The dentification of 13 novel variants, including four large deletions, was another highlight of our study, which emphasizes the genetic heterogeneity of MECP2 variants and underlines the need for generating population specific data. In view of identification of other recurrent variants in our study along with reported hot spot mutations, sequencing the MECP2 gene (beginning with exons 3 and 4) followed by MLPA testing if sequencing results are negative is recommended rather than targeted testing.
The majority of the variants were distributed in the functional domain of MECP2 with most missense variants clustered in MBD and truncating variants in ID and TRD of MECP2 [Figure 2], which support the findings of previous studies[3,7,8,16]. All variations identified in the patients were de novo. In one of the families, the mother was found carrying a different novel variant than her daughter and has no symptoms of the disease, as we reported previously[25].
Bioinformatic analysis revealed that most of the MECP2 missense variants clustered in MBD of MeCP2 were of damaging or deleterious nature, whereas all the missense variants identified in CTR were predicted as benign or non-deleterious. These findings support previously reported studies on RTT patients[26,27]. Only two missense variants were identified in TRD of MECP2 and bioinformatic analysis of the recurrent missense variant p.R306C in TRD predicted it as damaging, whereas the other non-recurrent missense variant p.T228S was predicted as benign or non-deleterious. The patient carrying this variant p.T228S was carrying another deleterious variant, p.P152R. As the number of missense variants identified in TRD in the present study was small, the effect of these variants could not be explored, but the findings of the present study strongly indicate that sequence variations in MECP2 gene are the major cause of classical RTT.
There are many studies on genotype–phenotype correlation in RTT patients from 2001 to 2016 but the results are inconsistent [16,18,21,27-32]. This inconsistency can be due to the use of different diagnostic criteria and severity scales for evaluation of the patients. There are currently no data on genotype-phenotype correlation in Indian patients with RTT. In this study, we tried to correlate the type and position of identified sequence variants with the phenotype of the patients.
When different types of sequence variations were compared with the phenotype, it was found that patients carrying early truncating variants showed more severe phenotype as compared to the patients with late truncating and missense sequence variants, supporting the findings from a previous study[18].
While comparing the types of sequence variations with their location in MECP2, it was found that the variants leading to severe phenotype were clustered more in functional domains of MeCP2. Only 5% of early truncating variants were present in MBD and CTR as compared to 20% present in the TRD. Only 5% of late truncating variants were observed in the CTR of MeCP2. The rare presence of missense variants in TRD or CTR of MeCP2 as compared to MBD can be explained on the basis that missense sequence variations may have mild impact on protein function compared to truncating sequence variations, resulting in a mild phenotype. These findings are in support of a previous study[26]. The only recurrent missense variant in TRD was p.R306C and the other missense sequence variations found in TRD and CTR (p.T228S, p.E394K, p.E397K, and p.P430S) were observed with single occurrence, supporting the previous hypothesis that most of the missense sequence variations within the TRD might be benign variants[26].
In conclusion, this study presents the largest cohort describing the molecular genetics of classical RTT from India. To the best of our knowledge, this is the first study showing the highest detection rate of MECP2 variants in the patients with classical RTT and supports that clinical stringency based on revised diagnostic criteria can increase the variant detection rate. We propose the following MECP2 screening strategy in Indian patients with Classical RTT. Exon 3 of MECP2 should be screened first, followed by exons 2 and 4 using Sanger sequencing, and, in turn, followed by quantitative analysis using MLPA. The present study adds information on the molecular characterization of Indian patients with RTT and also reports 13 novel variants expanding the genotypic spectrum of RTT. The findings can be useful for diagnostic testing, genetic counseling, and prenatal testing.
Limitations and future research
Although the study was performed on a large cohort of patients, we were unable to prove the functional impact of novel variations on the MECP2 protein as only software prediction tools were used. It would be useful to conduct functional studies for the new variants identified. A review of the current literature indicates that MECP2 variations can cause other neurodevelopmental phenotypes such as neonatal encephalopathy and atypical RTT phenotype in both males and females. This study lacks this information as only classical cases were included. A larger study is required to provide this information.
Declarations
AcknowledgmentsWe gratefully thank the patients and the families for their support and participation in this study. We thank Dr. Angus Clarke, Cardiff University for providing positive controls for common mutations. We thank Dr. R. Bandts (Maastricht University Medical Center) for providing technical support related to MLPA analysis. We thank Dr. Sumita Danda (Christian Medical College and Hospital, Vellore) and Dr. Veena Kalra (Indraprastha Apollo hospitals, New Delhi) for referring their patients to our institute.
Authors’ contributionsLiterature review, patient enrollment, experiment validation, planning and execution, data analysis and interpretation, bioinformatics analysis, wrote, prepared and reviewed manuscript: Khajuria R
Clinical evaluation, patient enrollment, counseling of families helped in planning the study, preparation and critical evaluation of the manuscript, reviewed the manuscript: Gupta N
Provided support with MLPA data interpretation and bioinformatics analysis using Alamut software, reviewed the manuscript: van Roozendaal KEP, Smeets EEJ, Curfs LMG
Helped in developmental assessment, behavioural evaluation and intervention for the patients, reviewed the manuscript: Sapra S
Helped in patient enrollent and counseling, reviewed the manuscript: Ghosh M
Helped in neurological evaluation and management of neurological co-morbidities and manuscript review: Gulati S
Conceptualized the study, helped in preparation of study protocol, helped in clinical evaluation, enrollment and counseling of patients, helped in preparation and critical evaluation of the study, reviewed of the manuscript, will act as the guaranteer for this study: Kabra M
Availability of data and materialsData source is from patients enrolled for the study. For data details corresponding author may be contacted.
Financial support and sponsorshipThis study was partially funded as research fellowship from Indian Council of Medical Research, New Delhi (Grant Number: 45/8/2006/Hum/BMS).
Conflicts of interestAll authors declared that there are no conflicts of interest
Ethical approval and consent to participateEthical approval for the present study was taken from the Ethics Committee of the Institute. Proper information about the study was given to all families and written informed consent was obtained from the parent/guardian.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Amir RE, Van den veyver IB, Wan M, Tran CQ, Francke U, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG –binding protein 2. Nat Genet 1999;23:185-8.
2. Ehrhart F, Coort SL, Cirillo E, Smeets E, Evelo CT, et al. Rett syndrome - biological pathways leading from MECP2 to disorder phenotypes. Orphanet J Rare Dis 2016;11:158.
3. Neul JL, Fang P, Barrish J, Lane J, Caeg EB, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008;70:1313-21.
4. Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010;68:944-50.
5. Percy AK, Neul JL, Glaze DG, Motil KJ, Skinner SA, et al. Rett syndrome diagnostic criteria: lessons from the Natural History Study. Ann Neurol 2010;68:951-5.
6. Hardwick SA, Reuter K, Williamson SL, Vasudevan V, Donald J, et al. Delineation of large deletions of the MECP2 gene in Rett syndrome patients, including a familial case with a male proband. Eur J Hum Genet 2007;15:1218-29.
7. Philippe C, Villard L, De Roux N, Raynaud M, Bonnefond JP, et al. Spectrum and distribution of MECP2 mutations in 424 Rett syndrome patients: a molecular update. Eur J Med Genet 2006;49:9-18.
8. Das DK, Raha S, Sanghavi D, Maitra A, Udani V. Spectrum of MECP2 gene mutations in a cohort of Indian patients with Rett syndrome: report of two novel mutations. Gene 2013;515:78-83.
9. Lallar M, Rai A, Srivastava P, Mandal K, Gupta N, et al. Molecular testing of MECP2 gene in rett syndrome phenotypes in Indian girls. Indian Pediatr 2018;55:474-7.
10. den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet 2001;109:121-4.
11. Christodoulou J, Grimm A, Maher T, Bennetts B. RettBASE: the IRSA MECP2 variation database-a new mutation database in evolution. Hum Mutat 2003;21:466-72.
12. Wan M, Lee SS, Zhang X, Houwink-Manville I, Song HR, et al. Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am J Hum Genet 1999;65:1520-9.
13. Erlandson A, Samuelsson L, Hagberg B, Kyllerman M, Vujic M, et al. Multiplex ligation-dependent probe amplification (MLPA) detects large deletions in the MECP2 gene of Swedish Rett syndrome patients. Genet Test 2003;7:329-32.
14. Archer HL, Whatley SD, Evans JC, Ravine D, Huppke P, et al. Gross rearrangements of the MECP2 gene are found in both classical and atypical Rett syndrome patients. J Med Genet 2006;43:451-6.
15. Khajuria R, Sapra S, Ghosh M, Gupta N, Gulati S, et al. Rapid detection of deletions in hotspot C-terminal segment region of MECP2 by routine PCR method: report of two classical Rett syndrome patients of Indian origin. Genet Test Mol Biomarkers 2009;13:277-80.
16. Bebbington A, Anderson A, Ravine D, Fyfe S, Pineda M, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology 2008;70:868-75.
17. Smeets E, Terhal P, Casaer P, Peters A, Midro A, et al. Rett syndrome in females with CTS hot spot deletions: a disorder profile. Am J Med Genet A 2005;132A:117-20.
18. Charman T, Neilson TC, Mash V, Archer H, Gardiner MT, et al. Dimensional phenotypic analysis and functional categorisation of mutations reveal novel genotype-phenotype associations in Rett syndrome. Eur J Hum Genet 2005;13:1121-30.
19. Yamada Y, Miura K, Kumagai T, Hayakawa C, Miyazaki S, et al. Molecular analysis of Japanese patients with Rett syndrome: Identification of five novel mutations and genotype-phenotype correlation. Hum Mutat 2001;18:253.
20. Pan H, Wang YP, Bao XH, Meng HD, Zhang Y, et al. MECP2 gene mutation analysis in Chinese patients with Rett syndrome. Eur J Hum Genet 2002;10:484-6.
21. Fukuda T, Yamashita Y, Nagamitsu S, Miyamoto K, Jin JJ, et al. Methyl-CpG binding protein 2 gene (MECP2) variations in Japanese patients with Rett syndrome: pathological mutations and polymorphisms. Brain Dev 2005;27:211-7.
22. Kim IJ, Kim YJ, Son BH, Nam SO, Kang HC, et al. Diagnostic mutational analysis of MECP2 in Korean patients with Rett syndrome. Exp Mol Med 2006;38:119-25.
23. Wong VC, Li SY. Rett syndrome: prevalence among Chinese and a comparison of MECP2 mutations of classic Rett syndrome with other neurodevelopmental disorders. J Child Neurol 2007;22:1397-400.
24. Gauthier J, de Amorim G, Mnatzakanian GN, Saunders C, Vincent JB, et al. Clinical stringency greatly improves mutation detection in Rett syndrome. Can J Neurol Sci 2005;32:321-6.
25. Khajuria R, Gupta N, Sapra S, Gulati S, Ghosh M, et al. Novel non-identical MECP2 mutations in Rett syndrome family: a rare presentation. Brain Dev 2012;34:28-31.
26. Yusufzai TM, Wolffe AP. Functional consequences of Rett syndrome mutations on human MeCP2. Nucleic Acids Res 2000;28:4172-9.
27. Weaving LS, Williamson SL, Bennetts B, Davis M, Ellaway CJ, et al. Effects of MECP2 mutation type, location and X-inactivation in modulating Rett syndrome phenotype. Am J Med Genet A 2003;118A:103-14.
28. Colvin L, Leonard H, de Klerk N, Davis M, Weaving L, et al. Refining the phenotype of common mutations in Rett syndrome. J Med Genet 2004;41:25-30.
29. Leonard H, Moore H, Carey M, Fyfe S, Hall S, et al. Genotype and early development in Rett syndrome: the value of international data. Brain Dev 2005;27:S59-68.
30. Robertson L, Hall SE, Jacoby P, Ellaway C, de Klerk N, et al. The association between behavior and genotype in Rett syndrome using the Australian Rett Syndrome Database. Am J Med Genet B Neuropsychiatr Genet 2006;141B:177-83.
31. Halbach NSJ, Smeets EEJ, van den Braak N, van Roozendaal KE, Blok RM, et al. Genotype-phenotype relationships as prognosticators in Rett syndrome should be handled with care in clinical practice. Am J Med Genet A 2012;158A:340-50.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Khajuria R, Gupta N, van Roozendaal KEP, Sapra S, Ghosh M, Gulati S, Smeets EEJ, Curfs LMG, Kabra M. Spectrum of MECP2 mutations in Indian females with Rett Syndrome - a large cohort study. J Transl Genet Genom 2020;4:91-103. http://dx.doi.org/10.20517/jtgg.2020.06
AMA Style
Khajuria R, Gupta N, van Roozendaal KEP, Sapra S, Ghosh M, Gulati S, Smeets EEJ, Curfs LMG, Kabra M. Spectrum of MECP2 mutations in Indian females with Rett Syndrome - a large cohort study. Journal of Translational Genetics and Genomics. 2020; 4(2): 91-103. http://dx.doi.org/10.20517/jtgg.2020.06
Chicago/Turabian Style
Khajuria, Rajni, Neerja Gupta, Kees E. P. van Roozendaal, Savita Sapra, Manju Ghosh, Sheffali Gulati, Eric E. J. Smeets, Leopold M. G. Curfs, Madhulika Kabra. 2020. "Spectrum of MECP2 mutations in Indian females with Rett Syndrome - a large cohort study" Journal of Translational Genetics and Genomics. 4, no.2: 91-103. http://dx.doi.org/10.20517/jtgg.2020.06
ACS Style
Khajuria, R.; Gupta N.; van Roozendaal KEP.; Sapra S.; Ghosh M.; Gulati S.; Smeets EEJ.; Curfs LMG.; Kabra M. Spectrum of MECP2 mutations in Indian females with Rett Syndrome - a large cohort study. J. Transl. Genet. Genom. 2020, 4, 91-103. http://dx.doi.org/10.20517/jtgg.2020.06
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 21 clicks
Cite This Article 21 clicks

 Like This Article 28
likes
Like This Article 28
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.