
Assigning single clinical features to their disease-locus in large deletions: the example of chromosome 1q23-25 deletion syndrome

Abstract
Aim: Assigning a disease-locus within the shortest regions of overlap (SRO) shared by deleted/duplicated subjects presenting this disease is a robust mapping approach, although the presence of different malformation traits and their attendance only in a part of the affected subjects can hinder the interpretation. To overcome the problem of incomplete penetrance, we developed an algorithm that we applied to the deletion region 1q23.3-q25, which contains three SROs, each contributing to the abnormal phenotype without clearly distinguishing between the different malformations. We describe six new subjects, including a healthy father and his daughter, with 1q23.3-q25 deletion of different sizes. The aim of this study was to correlate specific abnormal traits to the haploinsufficiency of specific gene/putative regulatory elements.
Methods: Merging cases with those in the literature, we considered four traits, namely intellectual disability (ID), microcephaly, short-hands/feet, and brachydactyly, and conceived a mathematical model to predict with what probability the haploinsufficiency of a specific portion of the deletion region is associated with one of the four malformations.
Results: The haploinsufficiency of PBX1 is strongly associated with ID. DNM3 and LHX4 are confirmed as responsible for growth retardation, whereas ATPIB1 was identified as a new candidate gene for microcephaly, short-hands/feet, and brachydactyly.
Conclusion: Although our model is hampered by long-term position effects of regulatory elements, synergistic cooperation of several genes, and incomplete clinical assessment, it can be useful for contiguous gene syndromes showing a complex pattern of clinical characteristics. Obviously, functional approaches are needed to warrant its reliability.
Keywords
Introduction
The usual method to identify the shortest regions of overlap (SRO) in contiguous gene syndromes relies on the graphical identification of the area of minimal overlap between deletions in patients sharing the same phenotype. Although this approach is very efficient when dealing with traits present in all the subjects who share the deletion region, it is much less productive when the trait is shared by some of the patients only. The usual way to overcome this uncertain correlation is to attribute an incomplete penetrance to the trait, a definition that may hide multiple factors such as the influence of any other genetic factors necessary for the manifestation of the trait, the differences in the breakpoints of the deletion involving any different dynamics of chromatin interactions between enhancers and promoters, environmental factors, or more simply inaccurate assignment of phenotype. Obviously, “non-penetrant” deletions may either overlap the disease locus (DL) or not include it, so that they constitute a limitation to defining SRO boundaries. However, they still strongly modulate the probability profile of the DL location along the SRO, i.e., the probability for the DL to map at a given position, considering the whole body of experimental data (i.e., all the deletions, either penetrant or non-penetrant, overlapping a given genomic position inside the SRO). In fact, the trait(s) considered in a given genomic region are often de novo and present in restricted numbers of subjects, so that the exclusion of even a single case can really be limiting to a correct locus assignment. Therefore, it is highly desirable to find a probabilistic model that, by considering also the “non-penetrant” cases, makes more reliable the assignment of specific traits to specific genomic portions. For this purpose, we propose a new genotype-phenotype correlation approach, applying our statistical procedure to interstitial deletions of 1q23.3-q25, of which more than 30 cases have been reported, with the imbalance being mainly de novo with the exception of three subjects who have inherited the deletion from the affected mother (Patients P10 and P17[1], Patient A[2], and Case 1[3]). These deletions are associated with a complex malformation condition consisting in proportionate pre- and postnatal growth deficit, cardiac malformations, small hands and feet with brachydactyly, intellectual disability (ID) of various degrees, and craniofacial dysmorphisms such as microcephaly, micrognathia, short nose with bulbous tip, dysplastic ears, elongated upper lip, and small chin have been reported in most subjects[1,3]. The relationship between the size and localization of the copy number variants and phenotypic abnormalities in thirty-five patients[1-9] allowed identifying three non-overlapping regions whose haploinsufficiency seemed crucial for the manifestation of some specific characteristics[1]. The SRO associated with growth and developmental delay has been progressively narrowed from 1.9 Mb[4] to a 179-kb region (chr1:172,460,683-72,281,412 hg19)[2]. A subregion of 2.5 Mb (chr1:164,501,003-167,022,133 hg19), located proximally to the SRO (SRO-P), adds further complexity to the observed phenotype, being more commonly associated with cardiac and renal malformations. A third distal region of 2.7 Mb (chr1: 178.514.910-181.269.712 hg19; SRO-D) could also contribute to intrauterine and postnatal growth retardation[1]. Finally, deletions involving SERPINC1 (chr1: 173.872.942-173.886.516; MIM: 107300) result in low antithrombin-III activity, a risk factor for thrombophilia. We present the detailed phenotypic and molecular description of six new cases whose partially overlapping 1q24q25 deletions were identified by chromosome microarray analysis (CMA). Four cases, each encompassing at least one of the three critical regions, were sporadic and identified by studying unrelated patients with syndromic intellectual disability. The fifth case, with a 1q24.3q25.2 deletion that did not involve any of the three critical regions, was ascertained in a newborn baby after the unexpected detection of the same deletion in the healthy father. The latter was studied as the parent of a previous child carrying two CNVs, neither of them located on chromosome 1, which later turned out to be both inherited from the healthy mother.
Methods
Clinical data
All six subjects are described in more detail in the following section and their clinical characteristics are summarized in Table 1. Patient photographs are shown in cases where parents have consented to publication.
Clinical features of the six subjects with 1q deletion
| Sporadic cases | Family | |||||
|---|---|---|---|---|---|---|
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | |
| Genotype | ||||||
| Chromosome bands | 1q23.1q24.2 | 1q23.3q24.2 | 1q24.2q25.3 | 1q24.3q31.3 | 1q24.3q25.2 | 1q24.3q25.2 |
| 180k CGH array: deleted interval, hg19) | chr1:164358165-168556004 | chr1:166325047-176709133 | chr1:170500104-184283033 | Chr1: 172579926-195051375 | chr1:172667560-178548677 | chr1:172667560-178548677 |
| Size of the deletion | 4.1 Mb | 10.3 Mb | 13.7 Mb | 22.5 Mb | 5.9 Mb | 5.9 Mb |
| Critical region involved* | SRO-P | SRO-P, 2, -I | SRO-I, -D | SRO-I, -D | None | None |
| Inheritance | De novo | De novo | De novo | De novo | De novo | Pat inherited |
| Parental origin | Not tested | Mat | Pat | Not tested | Pat | Pat |
| Phenotype | ||||||
| Age at diagnosis | 17 years | 4 years | 7 years | 4 years | 38 years | At birth |
| Sex | M | M | M | M | M | F |
| Mother’s age at birth | 29 years | 40 years | 18 years | 34 years | 31 years | 31 years |
| Father’s age at birth | 31 years | 43 years | 20 years | 34 years | 37 years | 38 years |
| Previous clinical suspicion | None | Aarskog-Scott and Robinow Syndromes | Aarkog-Scott and Robinow Syndromes | None | None | |
| Previous negative genetic testing | K, Tel-MPLA, Frax-A/-E | K, Tel-Fish, FDG1, GJB-WSI** gene variants | K, FDG1 gene variant | K | None | None |
| Perinatal | ||||||
| IUGR | No | Yes | Yes | Yes | No | Yes |
| Week of gestation | At term | 40 weeks | 36 weeks | 43 weeks | NR | 37 weeks |
| Birth weight | 50th percentile | 3 rd percentile | 3rd percentile | -3 SD | NR | 3rd percentile |
| Birth OFC | 25th percentile | -2 SD | 3rd percentile | NR | NR | -2 DS |
| Birth length | 50-75th percentile | -1.75 SD | 3rd percentile | NR | NR | 10th percentile |
| Breast feeding difficulties | Yes | No | NR | NR | No | Yes |
| Post-natal growth delay | No | Yes | Yes | Yes | No | No |
| At last evaluation | ||||||
| Age | 18 years | 5 years | 7 years | 10 years | 38 years | 8 months |
| Weight | 10-25th percentile | -2 SD | << 3rd percentile | < 10th | 25th percentile | 10-25th percentile |
| OFC | 25-50th percentile | -2 SD | -4 SD | -3 SD | 25th percentile | -3 SD |
| Height | ≤25th percentile | -3.5 SD | -4 SD | -2 SD | 25th percentile | 50-75th percentile |
| Neurodevelopment and CNS | ||||||
| Intellectual disability (degree) | Moderate (IQ:54) | Moderate (IQ:50) | Severe (IQ:38) | Severe (IQ:44) | No | NA |
| Delayed/absent speech | Coprolalia and soliloquy | Absent | Absent | Absent | No | NA |
| Hypotonia | Yes | No | Yes with joint laxity | No | No | |
| Epilepsy/seizures | No | + (Focal epilepsy) | Non-epileptic myoclonus | No | No | No |
| Sensorineural hearing loss | No | + (mild) | No | NR | No | NT |
| Brain MRI | Normal | Enlarged ventricles | NT | Thin corpus callosum | NT | NT |
| Behavior | Repetitive movements, OCT, apathy, abulia | Aggressiveness, hyperactivity | Impulsiveness | None | ||
| Facial dysmorphisms | ||||||
| Sparse hair/eyebrow | No | Yes | Yes | Yes | No | No |
| Hypertelorism | No | Yes | Yes | No | No | |
| Dysplastic ears | Yes | Yes | Yes | Yes | No | No |
| Bulbous/broad nose | No | Yes | Yes | Yes | No | |
| Abnormal lip/palate | No | Yes | Yes | Yes | No | No |
| Micro/retrognathia | No | Yes | Macrostomia | Yes | No | Yes |
| Short/broad neck | Broad neck/slight pterigium colli | Yes | Yes | No | Short neck | |
| Hands/feet | ||||||
| Short | No | Yes | Yes | Yes | Yes | No |
| Brachydactyly | No | Yes | Yes | Yes | No | No |
| 5th finger clinodactyly | No | Yes | Yes | Yes | No | No |
| Broad thumbs/halluces | No | Yes | Yes | No | No | No |
| Single palmar crease | No | Yes | Yes | No | No | No |
| Fingers hyperextensibility | Yes | Yes | + (mild) | No | ||
| Malformations | ||||||
| Genitalia | No | Micropenis | Unilateral cryptorchidism | Unilateral | No | |
| Kidney | No | NT | Normal | No | No | |
| Heart | No | No | No | Mild mitral insufficiency | No | No |
| Skeleton | Spinal kyphosis and decreased lumbar lordosis | Delayed bone age | NT | NT | ||
| Miscellaneous | Supraclavicular cyst of cartilage | |||||
Short regions of overlap identified in this study
| SRO | Chromosome band | Coordinates [hg19] | Size | Phenotypic trait | Candidate gene |
|---|---|---|---|---|---|
| SRO-1 | 1q23.3 | 164761881- 164799811 | 38 kb | ID | PBX1 (ex 4-8) |
| SRO-2 | 1q24.3-24.3 | 168556004- 170516641 | 1.9 Mb | MCH/SKA | ATP1B1 |
| SRO-3 | 1q25.2 | 177238295- 178548677 | 1.3 Mb | ID/MCH | FAM5B |
Case 1
The patient was a 17-year-old male born after a previous miscarriage. At birth, his mother and father were 29 and 31 years old, respectively. He has a younger healthy brother. Family history was remarkable for cognitive delay in the paternal lineage, not otherwise specified, and bipolar disturbance in the maternal one. The delivery was at term with fetal distress consisting of decreased heart rate patterns and meconium-stained amniotic fluid. Birth weight was 3,450 g (50th percentile), length was 51 cm (50th percentile), and cranial circumference-OFC was 35 cm (25th percentile). Apgar scores were 8/9 at 1’/5’, respectively. The perinatal period was remarkable for hypotonia and limb dyskinesia. Early motor milestones were slightly delayed: he sat between 7 and 8 months, crawled at 12 months, and walked autonomously at 18 months. Language and learning difficulties were noticed early. He started babbling at 18 months and language development was delayed. At the age of 13 years, Griffiths scale scores revealed moderate ID (overall IQ: 54) with pragmatic and narrative language difficulties and social skills impairment. Physical examination revealed craniofacial dysmorphisms including short neck with slight pterigium colli, hypoplasia of auricles with flat helix. His OFC was 53.8 cm (25th-50th percentile). In addition, ligamentous laxity, hyperextensibility of the finger joints, bilateral flat foot with sandal gap, bilateral genu valgus, spinal kyphosis, and decreased lumbar lordosis were observed. A supraclavicular cartilage cyst (3-mm diameter) was noted. Brain magnetic resonance imaging, kidney ultrasound, urinalysis, and functional blood tests were normal. The thyroid function test gave normal results. At the age of 16 years, he was classified as suffering from severe intellectual disability with marked repetitive movements, obsessive-compulsive traits, apathy, and abulia with episodes of coprolalia and soliloquy, without any self-hetero-aggressive behavior. Treatment with Risperidal or Abilify was recommended. At the age of 182/12 years, his height was 171 cm (< 5th percentile), weight 59.7 kg (10th-25th percentile), and OFC was 55.4 cm (25th-50th percentile). Array-CGH revealed a 4.2-Mb deletion of 1q23.3q24.2.
Case 2
The male child was born to a 40-year-old primigravida and her 43-year-old partner. Due to the father’s oligospermia, the couple underwent two cycles of in vitro fertilization (IVF) through ICSI (intracytoplasmic sperm injection), which led to the conception of the patient. The delivery was normal after an unremarkable 40-week pregnancy with a birth weight of 2,380 g (third percentile), length of 45 cm (-2 SD), and cranial circumference-OFC of 33 cm (-1.75 SD). Apgar scores were 10/10 at 1’/5’, respectively. At 5.5 months, he began experiencing recurrent episodes of non-febrile seizures when falling asleep or waking up. Electroencephalogram (EEG) recording showed bilateral and rare paroxysmal slow abnormalities in the fronto-temporal region. A therapy with levetiracetam achieved a reduction of seizure frequency. At age 9.5 months, his height was -2.5 SD. At clinical examination, facial dysmorphisms including prominent forehead, hypertelorism, saddle nose, micrognathia, smooth philtrum with vermillion upper lip, small ears with hypoplastic helix, slight neck pterygium, sparse hair [Figure 1A], and micropenis were observed. Audiological testing revealed mild sensorineural hearing impairment. At the age of 16 months, he started walking alone and expressive speech was absent. At the age of 22 months, his height was 74 cm (-3 SD), weight was 10 kg (-2 SD), and OFC was 47 cm (-2 SD). His hands and feet were broad with brachydactyly [Figure 2A]. X-ray showed delayed bone age of 1.3 years. EEG displayed focal epileptiform abnormalities: spike-wave complexes on the left hemisphere. Patient was stable on the therapy with levetiracetam (2 cp × 90 mg). The brain MRI showed an enlarged third ventricle. Aarskog-Scott syndrome (OMIM 305400) was excluded following normal results of FDG1 gene mutation analysis. By 4.5 years of age, his height had decreased to -3.5 SD. His thyroid function and insulin-like growth factor-1 (IGF-1) level were normal. At the last evaluation at the age of five years, Griffiths scale scores revealed moderate ID (IQ: 50), language was absent, and the previously friendly behavior was now characterized by aggression and hyperactivity. Karyotype was normal and array-CGH revealed a 10.3-Mb deletion of 1q24.1q25.2 [Supplementary Figure 1A].
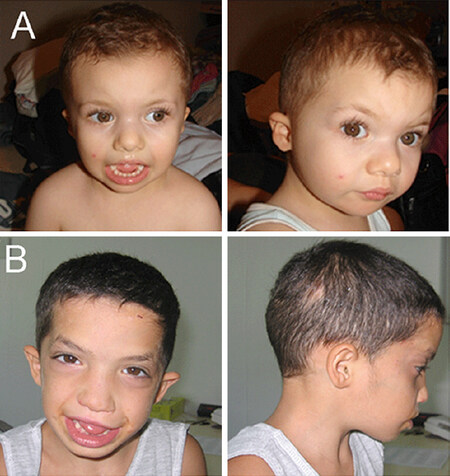
Figure 1. Front and lateral views of individuals with the 1q deletion. On facial profile Case 2, aged three years (A) and Case 3, aged seven years (B), ears appear low-set and posteriorly rotated with mild malar hypoplasia and microcephaly
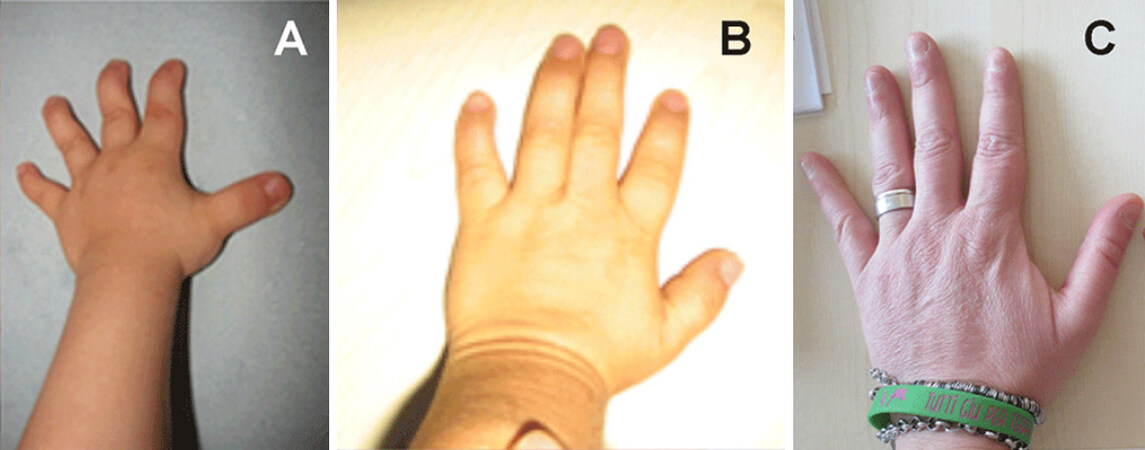
Figure 2. Hands of subjects with the 1q deletion. Small hands with distinctive brachydactyly, single transverse palm creases and fifth-finger clinodactyly are evident in Case 2 (A) and Case 3 (B). In contrast, the hands of Case 5 appear normal apart from mild hyperlaxity (C)
Case 3
The patient was born to an 18-year-old mother and a 20-year-old father, after a pregnancy characterized by the risk of miscarriage. He was delivered vaginally at 36 weeks with weight, length, and OFC far below third percentile. He was admitted for prematurity to Neonatal Intensive Care Unit. Peculiar dysmorphic features and hypotonia were noted. A diagnosis of Aarskog syndrome was suggested but not confirmed by the molecular analysis of FDG1gene. He had a normal karyotype, 46,XY.
His medical history was positive for failure to thrive and psychomotor delay. He started to walk unsupported at the age of three years and never developed verbal language. At the age of four years, he underwent surgical correction for unilateral cryptorchidism. At the last evaluation at the age of seven years, he showed severe psychomotor delay. His weight was 15 kg (< 3rd percentile), height was 99 cm (-4 SD), and OFC was 44 cm (-4.2 SD). Physical examination revealed high frontal hairline, down-slanting palpebral fissures, hypertelorism, depressed nasal bridge, mild malar hypoplasia, anteverted ears, deep philtrum, and macrostomia [Figure 1B]. His hands were small with short fingers, bilateral clinodactyly of the fifth finger, and bilateral single palmar crease. His feet were small with short toes, broad hallux, and bilateral “sandal gap” [Figure 2B]. Other findings included mild hypotonia and joint laxity. Griffiths scale scores revealed severe ID (IQ: 34; Performance: 34) with absent language. EEG recordings showed an excess of fast rhythms particularly over anterior areas. During sleep, bursts of paroxysmal slow abnormalities were present bilateral, diffuse, and prevalent in anterior left areas. Non-epileptic myoclonus was present both during wakefulness and sleep. His behavior was characterized by impulsiveness. Array-CGH revealed a 13.7-Mb deletion of 1q24.2q25.3 [Supplementary Figure 1B].
Case 4
The 10-year-old patient was the first child born to 34-year-old healthy, non-consanguineous parents. Familiarity for cleft lip/palate and deaf-mutism was recorded. He has a younger healthy nine-year-old brother. The patient was delivered by caesarian section at 43 weeks of gestation after a pregnancy characterized by IUGR and poor fetal movements. His birth weight was 2,900 g (-3 SD); length and OFC were not recorded. Cleft lip/palate was surgically corrected at the age of one year. At the age of two years, his psychomotor and language development was moderately delayed and characterized by inattentive-hyperactive behavior. At the same age, left-side cryptorchidism was surgically corrected. Mild growth hormone deficiency was documented but without the need for pharmacological treatment. When evaluated at the age of 10 years, his weight was 24 kg (< 10th percentile), height was 120 cm (3rd-10th percentile), and OFC was 49 cm (-3 SD). At neurologic examination, he presented global clumsiness and facial dysmorphic features including triangular face, highly arched eyebrows, upslanting palpebral fissures, high nasal bridge, short phyltrum, cleft lip and palate with malposition of incisors, arched palate, micrognathia, and low-set ears. Mild calcaneovalgus foot was also noted. Leiter-R scores revealed moderate ID (IQ: 44) with absent speech. ECG revealed mild mitral valve insufficiency. Brain MRI showed hypoplasia of the corpus callosum. Array-CGH revealed a 22.4-Mb deletion of 1q24.2q25.3 [Supplementary Figure 1B].
Cases 5 and 6
The pedigree is shown in Figure 3.
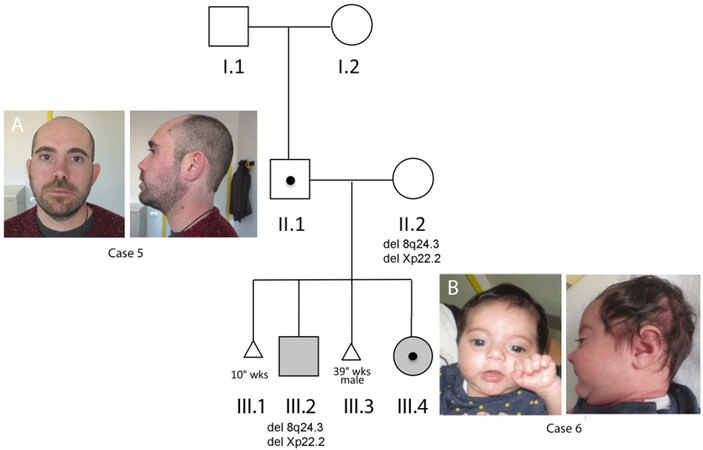
Figure 3. Pedigree of family, shaded square and circle: clinically affected. Front and lateral views of Case 5 (A) and Case 6 (B). Black dot: 1q24.3q25 deletion carrier
Case 5 (II.1) and Case 6 (III.4) are with 1q24.3q25 deletion, while III.2 carries two microdeletions at 8q24 and Xp22.2 (see Supplementary Figure 2 for details), both maternally inherited (II:2). In paternal grandparents (I.1 and I.2), the presence of 1q deletion was excluded by CMA.
Front and lateral views of Case 5 (A) and Case 6 (B). While Case 5, at the age of 38 years, does not show any of the facial dimorphisms reported in patients with overlapping 1q deletion, his daughter (Case 6), at the age of nine months, showed thin upper lip, microretrognathia, short neck, and microcephaly.
The index patient (III.2) was a four-year-old child born to a 27-year-old mother, who during pregnancy suffered from preeclampsia and was treated with anticoagulant drugs (aspirin and heparin) for thrombophilia and eutirox for hypothyroidism. The delivery was induced at 36 weeks of gestation for oligohydramnios. His birth weight was 2,800 g (10th-50th percentile). The perinatal and neonatal period was unremarkable despite feeding difficulties characterized by gastroesophageal reflux until the age of nine months. He crawled at 10 months and walked alone at the age of 18 months. His speech development was delayed and, at the age of four years, he was able to pronounce incomplete words. His behavior was characterized by low frustration tolerance associated with heteroaggressivity and bruxism. A diagnosis of autism spectrum disorder (ASD) was made (QS: 75, F 84.0 ICD 10, 299.00 ICD 9). CMA revealed two deletions at chromosomes 8q24.3 of 124 kb and Xp22.2 of 58.9 kb [Supplementary Figure 2], both inherited from the healthy mother.
Subject II.1 (Case 5) was a 38-year-old adult male of Sardinian origin. The CMA, which was performed to investigate whether the CNVs on chromosomes 8 and X [Supplementary Figure 2] detected in his son (Subject III.2) were de novo or inherited, revealed a deletion at 1q24.3q25.2 of 5.8 Mb later proved to be de novo[Supplementary Figure 1]. Careful clinical examination revealed a healthy man without any facial dysmorphisms [Figure 1C] or other abnormal features but for mild fingers ligamentous hyperlaxity at the hands [Figure 2C]. His height was 164 cm, at the 25th percentile for the Sardinian population[10], and his cranial circumference-OFC was 54 cm (25th percentile). The 1q24.3q25.2 deletion was established while his wife (Subject II.2) was 27 weeks pregnant (Subject III: 4, Case 6) and undergoing therapy for gestational diabetes and platelet aggregation inhibitors due to a previous miscarriage (III.1) at the 10th week of pregnancy and a subsequent intrauterine fetal death at the 39th weeks (III.3). This stillborn male was of 2,850 g (10th percentile), the cranial circumference-OFC of 29 cm (-3 SD) and length of 45 cm (< 3rd percentile). The morphological examination did not reveal any congenital malformation, while autoptic microscopic observation revealed macerated internal organs and venous thrombosis of umbilical cord, leading to a diagnosis of IUFD consistent with mild-moderate chorioamnionitis and fetoplacental thrombotic vasculopathy. DNA analysis was not performed. CMA on mother’s blood revealed two deletions at chromosomes 8q24.3 of 124 kb and Xp22.2 of 58.9 kb [Supplementary Figure 2].
Patient III.4 (Case 6) was a female delivered by caesarian section at 37 weeks of gestation because of growth retardation (IUGR) and poor fetal movements. Her birth weight was 2,170 g (3rd percentile), length was 45 cm (10th percentile) and cranial circumference-OFC was 30 cm (-2 SD). Apgar scores were 10/10 at 1’/5’, respectively. The perinatal period was unremarkable, although, due to her inability to attach to the breast, she was fed on infant formula. The first neuropediatric assessment occurred at three months of age, showing OFC parameters of 35.2 cm (-3 SD), weight of 4,600 g (25th percentile), and length of 58 cm (50th-75th percentile). At the same age, cerebral ultrasound gave normal results. At last evaluation at the age of 8 months, her OFC was 39 cm (-3 SD) and weight was 7.5 kg (10th-25th). Minor facial dysmorphisms were noted [Figure 1D]. CMA analysis, performed at birth in light of her father’s CMA finding, highlighted the same 1q24.3q25.2 deletion of 5.8 Mb [Supplementary Figure 1D]. At the age of 41 days, routine chromogenic plasma testing revealed low antithrombin activity 3 (32%, normal 80%-120%), similar to what was documented in the father (45%, normal 70%-130%)[3].
Molecular investigations
After obtaining the informed consent approved by the ethics committee for research at the corresponding institutions, DNA samples were prepared from blood of all six subjects and their parents. The study was conducted in accordance with the Declaration of Helsinki and national guidelines.
Chromosomal microarray analysis
CMA was performed on all six subjects and their parents using Agilent Human Genome CGH Microarray Kit 4 × 180 k (G4449A) and CGH-SNP array (G4890A) in order to investigate the parental origin of the CNV and whether it was de novo or inherited.
Data analysis was done using Agilent Cytogenomics V.2.5.8.1. All nucleotide and SNP positions refer to the Human Genome, February 2009 Assembly (hg19). Molecular karyotype was designed according with the International System for Human Cytogenetic Nomenclature (ISCN 2016).
Parent of origin determination
We genotyped family trios in three subjects (Cases 2, 3, and 6) by CGH-SNP array (Agilent 4 × 180k).
Gene content analysis
The gene content for each SRO was analyzed taking into account the haploinsufficiency (HI) and loss-of-function intolerance (pLI) scores. The HI score is defined as the predicted probability that a gene is more likely to exhibit haploinsufficiency (0%-10%) or more likely not to exhibit haploinsufficiency (90%-100%) based on differences in characteristics between known haploinsufficient and haplosufficient genes (https://decipher.sanger.ac.uk/).
The pLI score represents the probability that a gene is extremely intolerant of loss-of-function variation (pLI ≥ 0.9). Genes with low pLI scores (≤ 0.1) are loss-of-function tolerant. This score is based on protein-truncating variants in the GnomAD database (https://gnomad.broadinstitute.org/). Moreover, according to gnomAD Gene constraint suggestions, to evaluate highly likely haploinsufficient genes, we also used the observed/expected score.
Whole-exome sequencing analysis
Whole-blood samples of all available family members [Figure 3], except for the newborn baby (Case 6), were collected for WES analysis, which was performed by an external service provider (BGI Genomics, Hong Kong). According to the provider’s description, whole-exome enrichment was carried out using Illumina kit and sequenced with the DNB-SEQ500 to generate 100-bp-paired end reads that were aligned to the human genome (UCSC GRCh38), at an average coverage of 150 ×.
GATK tools (https://software.broadinstitute.org/gatk/)[11], including base quality score recalibration, indel realignment, duplicate removal, and SNP, and insertions/deletion (INDEL) identification were used according to the recommendations of GATK best practices[12]. Variants were annotated using the ANNOVAR software tool[13] and then filtered according to their predicted effects and allele frequencies in the available public databases (dbSNP http://www.ncbi.nlm.nih.gov/projects/SNP/, ExAC http://exac.broadinstitute.org/, 1000 Genomes http://www.1000genomes.org/, and ESP6500 http://evs.gs.washington.edu/EVS/).
Probability profiling of genomic regions linked to selected traits
To computationally infer the genomic segments being most likely associated with selected clinical features, we assumed that a specific trait was predominantly the outcome of the hemizygosity of specific DL, either a protein-coding gene or a putative regulatory element, rather than the synergistic effect of the haploinsufficiency of several genomic elements.
Given this assumption, the probability for a DL to map at a given genomic location essentially depends on the penetrance of its haploinsufficiency and on the causative and non-causative deletions that overlap the genomic position.
For a detailed description of the mathematical model, see the Supplementary Materials (Mathematical Model).
Briefly, molecular data from patients, in whom the clinical status for a specific trait was assessed, were grouped and analyzed independently. Clearly, as not all patients were evaluated for a specific trait, the number of individuals in each group varied. In the first step of the procedure, we identified SRO regions, taking into account only overlaps between deletions associated with the trait. By definition, these SROs have probability 1 to contain the DL. The next step was to estimate the probability distribution inside SRO(s). At this purpose, we used a Bayesian approach to calculate, for each non-overlapping sliding window (Δ) of 1 kb within the SRO, the posterior probability to intersect the DL, conditioned by the experimental data (i.e., all the deletions overlapping the specific window inside the SRO). In this regard, we assumed that the a priori probability P (Δ overlaps DL) was inversely proportional to the SRO size and that the best estimator for the penetrance of the DL was the value which maximizes the likelihood function P (Experimental data given that Δ overlaps DL) (see Supplementary Materials, Mathematical Model).
In the last phase of the procedure, for each clinical feature (intellectual disability, microcephaly, kidney malformations, dysplastic ears, hypertelorism, short hands and feet, hypotonia, brachydactyly, microretrognathia, speech delay, and walk delay), custom UCSC tracks were automatically built to visualize in their genomic context the set of deletions and the probability profiles, calculated either in absolute or in log-scale. The software is available on request.
Results
Chromosomal microarray analysis
The deletions were within bands 1q23.3-1q31.3 and ranged in size from 4.1 to 22.5 Mb [Figure 4]. One deletion (Case 6) case was inherited from unaffected father (Case 5, Figure 3).
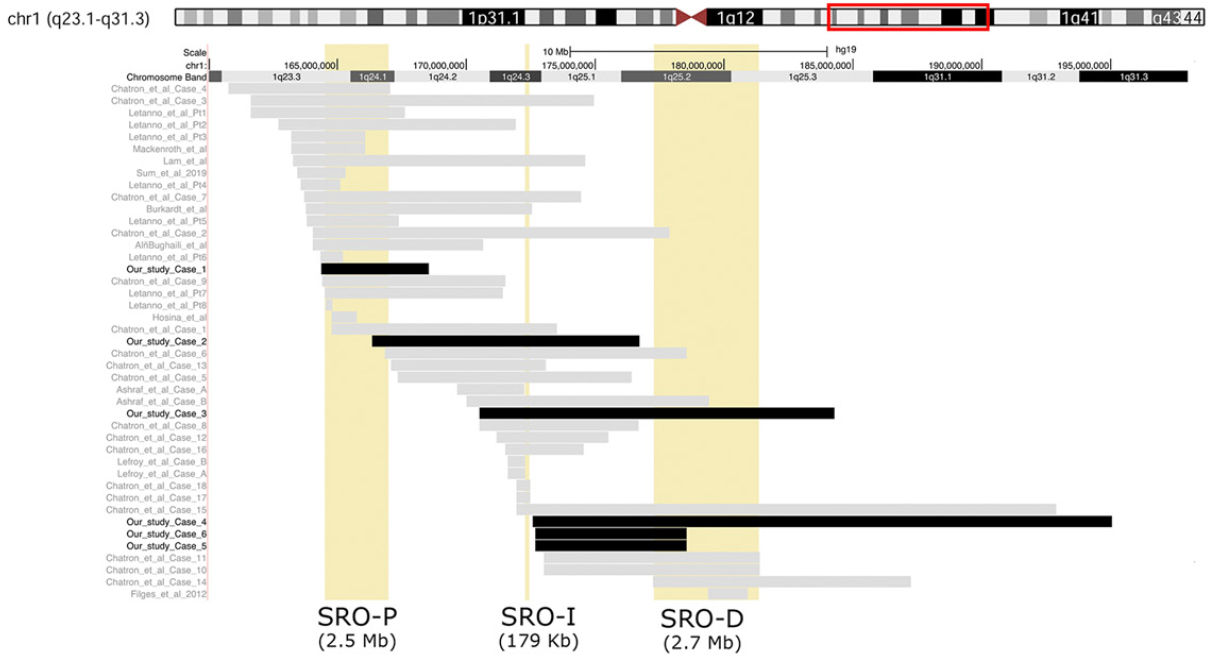
Figure 4. Overview of 1q23-q31 deletions. Alignment of the 1q deletions detected in our six cases (black bars) and literature patients (grey bars). The screenshot spans 31.8 Mb of chromosome 1q23.3-q31.3. The yellow box indicates the three short regions of overlap (SRO) previously defined[1,2]. The size of each SRO is indicated (see Discussion)
The chromosomal breakpoints defined by CMA [Supplementary Figure 1] in the six cases were:
Case 1:
arr[GRCh37] 1q23.3q24.1(164343729x2,164358165_168556004x1,168586532x2)dn
Case 2:
arr[GRCh37] 1q24.1q25.2(166270638x2,166325047_176709133x1,176724229x2)dn
Case 3:
arr[GRCh37] 1q24.2q25.3(170483844x2,170500104_184283033x1,184300213x2)dn
Case 4:
arr[GRCh37] 1q24.3q31.3(172571382x2,172579926_195051375x1,195099271x2)dn
Case 5 (Subject II.1, Figure 3):
arr[GRCh37]1q24.3q25.2(172643270x2,172667560_178548677x1,178565397x2)dn
Case 6 (Subject III.4, Figure 3):
arr[GRCh37]1q24.3q25.2(172643270x2,172667560_178548677x1,178565397x2)pat
Parental origin analysis
The parent of origin was determined for three subjects, two (Cases 3 and 5) paternal and one (Case 2) maternal [Supplementary Table 1]. The parental DNA samples of the remaining subjects (Cases 1 and 4) were unavailable.
Whole-exome sequencing
Exome sequencing of Subject II.1 and his parents (trio analysis) did not provide a strong candidate variant likely relevant for ASD. Taking advantage of whole-exome sequencing data on the mother, we ruled out the possibility that the paternally inherited 1q24.3q25.2 deletion on Patient III.4 might have unmasked a recessive allele lying on the maternal chromosome. We also explored the hypothesis that variants in genes involved in coagulation cascade or fibronolysis could cause inherited predisposition to thrombophilia, possibly linked to the recurrent miscarriages observed in the mother. Interestingly, while we did not identify any candidate variants in genes already associated with thrombophilia, WES analysis demonstrated in the mother two missense variants (NM_001061.6:c.796C > T:p.R266W and c.1279G > A:p.A427T) in the TXBAS1 gene (MIM 274180) [Supplementary Figure 3]. These variants were in trans as only one of them (c.796C > T) was identified in the son (Subject II.1), rare (AF < 0.001 on several databases), and predicted to impact on protein function by in silico analysis. Both variants were technically verified by Sanger sequencing. The TBXAS1 gene encodes the enzyme thromboxane synthase (TXAS), which catalyzes the conversion of prostaglandin H2 to thromboxane A2 (TXA2), a potent vasoconstrictor and inducer of platelet aggregation[14]. Biallelic missense mutations in TBXAS1, accounting for a decreased TXAS activity, have been associated with the Ghosal hematodiaphyseal syndrome (MIM 231095), a disease characterized by abnormal bone remodeling and anemia.
Short region of overlaps
According to the genotype-phenotype correlations emerging from our study, we defined three new SROs, each associated with specific phenotypic traits, such as ID, microcephaly (MCH), and skeletal anomalies including short hands and feet and brachydactyly. Their details are summarized in Table 2 and visualized in Figure 5.
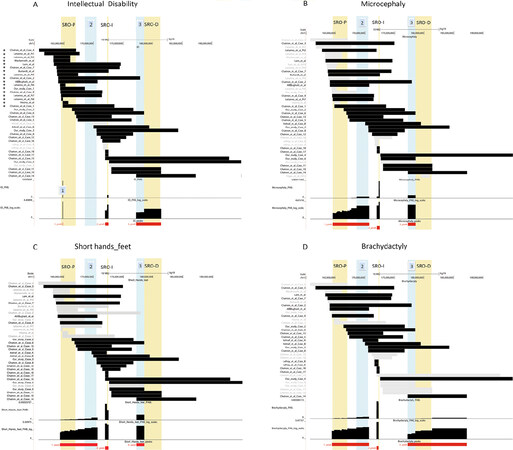
Figure 5. Visualization of the short region of overlaps (SROs) and probability profiling of genomic regions linked to selected traits. (A-D) (top) A screenshot covering 31.8 Mbs at 1q23.3-q31.3 shows the alignment of the deletions in our cases and in the literature. The black bars indicate cases with the selected clinical feature among those reported in del(1) cases [(A) intellectual disability, (B) microcephaly, (C) Short hands and feet, and (D) brachydactyly], while the gray bars represent cases without. The vertical yellow box indicates the proximal SRO (SRO-P, chr1:164,501,003-16,7022,133) of Chatron[1], while the light blue vertical box represents the new SROs indicated by 1-3 defined by this study (see Table 2 for the details). (bottom) A graph showing the estimated probability distribution of the genomic location of the disease loci associated with the traits
Probability profiling of genomic regions linked to selected traits
Computational prediction of the DLs localization allowed us to identify in the eleven traits examined a total of 26 SROs with sizes ranging from 0.038 to 22 Mb (mean: 3.5 Mb) [Supplementary Table 2].
Importantly, genomic intervals having a cumulative probability to contain the DL > 85% are considerably shorter than their corresponding SROs, reducing the number of high-priority candidate genes. Striking examples of this reduction concern SROs related, respectively, to ID (Peaks 1 and 2), brachydactyly (Peaks 1 and 2), and microcephaly (Peak 1) [Supplementary Table 2].
To check whether the intrinsic limitation of CMA to precisely map the breakpoints could affect the results, we decided to perform the analysis taking into account for each rearrangement either the smallest or the largest deletion regions, as defined by CMA. Interestingly, while nine out of eleven trait-related analyses remained unaffected, results concerning the traits “dysplastic ears” and “speech delay” markedly differed in the localization and size of the intermediate SRO [Supplementary Figures 4 and 5], essentially due to a different set of overlapping deletions involved in the definition of that SRO. This finding suggests that caution should be applied in drawing firm conclusions when interpreting the results, as uncertainty about exact breakpoint localization as well as inaccuracy of clinical assessment may lead to erroneous SRO localization and probability profiles.
Discussion
In this paper, we describe six individuals with deletions scattered within chromosome 1q23.1q25 where three specific SRO deletion syndromes are reported: a proximal one of 2.5 Mb (SRO-P, chr1:164,501,003-167,022,133)[1], an intermediate one of 170 kb (SRO-I, chr1:172,460,683-172,281,412)[2], and distal one of 2.7 Mb (SRO-D, chr1:177,238,295-181,400,912 )[1][Figure 4].
In particular, two SROs (SRO-I and -D) are described as associated with IUGR resulting in short stature up to -5 SD, microcephaly up to -4 SD, small hands and feet with fifth finger clino-brachydactyly, and variable degree of ID, in addition to peculiar facial dysmorphisms[1,2,4,6].
Indeed, our subjects with deletion including both SRO-I and -D (Figure 4, Cases 2-4) exhibited all these features [Figures 1 and 2, Table 1]. On the contrary, our Case 5 with a de novo 1q24.3q25.2 deletion (5.9 Mb, chr1:172,667,560-178,548,677) that did not contain the SRO-I and only partly the proximal portion of SRO-D [Figure 4] has no ID or microcephaly, works in a qualified profession, and has a stature at the 25th percentile for the Sardinian population[7]. He also does not show any of the dysmorphic features [Figure 3A and Table 1] reported for the overlapping 1q24q25 deletions. However, his nine-month-old daughter (Case 6) with the same 1q deletion [Figure 4] had a history of IUGR and showed mild craniofacial dysmorphisms, including microcephaly (-3 SD), micro-retrognathia, and short neck [Figure 3B], as observed in our Cases 2-4 and in patients reported with overlapping 1q deletions including both the intermediate and distal SROs [Table 1].
Figures 5 and 6 illustrate the new SROs we defined according to the genotype-phenotype correlations emerging from our study.
SRO-P (chr1:164,501,003-167,022,133) was narrowed from 2.7 Mb to 38 kb (chr1:164,761,881-164,799,811) within a region involving exons 4-8 of PBX1 that in Figure 6A and B is indicated as N.1.
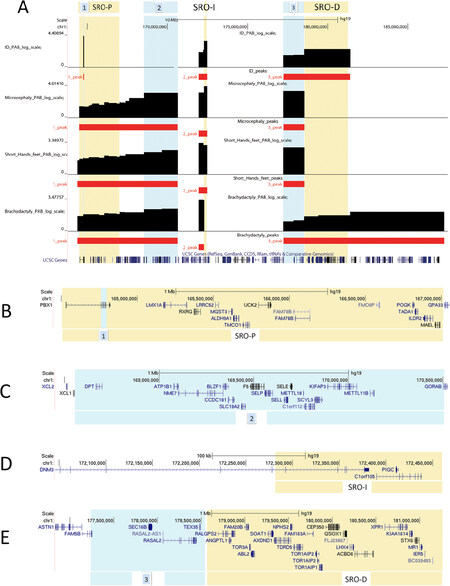
Figure 6. Overview of the short region of overlaps (SROs) and probability profiling of genomic regions linked to selected traits. A graph showing the estimated probability distribution of the genomic location of the DLs associated with the traits intellectual disability, microcephaly, short hands and feet, and brachydactyly (A); and magnified view of UCSC genes (GRCh37/hg19) mapping within the SROs (B-E)
This deletion is common to the 17 cases, including our Case 1, listed with an asterisk in Figure 5A with deletions ranging from 276 kb to 14.1 Mb. All have from severe to moderate ID but not microcephaly [Figure 5B]. The probability mass distribution at this region, as computationally calculated according to methods [Supplementary Table 2], shows that highest values for ID, kidney abnormalities, dysplastic ears, hypotonia, and speech delay [Figure 6A, Supplementary Figures 4 and 5].
Consistent with these findings, de novo, deleterious PBX1 sequence variants result in a highly variable syndromic form of intellectual disability, which includes external ear abnormalities and congenital defects of the kidney and urinary tract. In fact, most patients with 1q deletion including PBX1 have renal abnormalities and the association between Cakut syndrome and PBX1 variants/deletions is well demonstrated[15,16]. PBX1 alterations may also contribute to severe behavioral traits such as autism and obsessive-compulsive disorder[15]. Indeed, in addition to moderate ID, the phenotype of our Case 1 was characterized by repetitive movements and psychiatric traits resembling Tourette syndrome, such as obsessive-compulsive behavior with episodes of coprolalia and soliloquy [Table 1]. Interestingly, PBX1 gene has been recently identified among pleiotropic risk loci that play important roles in the neurological development processes associated with psychiatric disorders[17].
We also identified a 1.9-Mb genomic region (168,556,004-170,516,641) showing a high cumulative probability for microcephaly, brachydactyly, and short hands and feet [Figures 5 (N.2) and 6A (N.2)].
Within this region, the ATP1B1 gene [Figure 6C] shows the highest pLI value with observed/predicted scores indicating haploinsufficiency intolerance [Supplementary Table 3]. ATP1B1 (OMIM 182330) encodes for the subunit ß1 of Na,K-ATPase family, responsible for the homeostasis of the electrochemical gradients of Na and K ions across the plasma membrane. While mutations of the of Na,K-ATPase α-subunits have already been associated with neurological diseases (reviewed by Clausen et al.[18]), no confirmed mutations in any of the ß-subunits have yet been correlated with human disorders. Interestingly, ATP1B1, as part of an Na,K-ATPase multiprotein complex, interacts with the calcium channel TRPV4 (MIM *605427)[19] whose heterozygous de novo variants, either or missense, associate with skeletal disorders. Similar to our patients (Cases 2 and 3), those with TRPV4-related skeletal dysplasias include short stature, small hands and feet, and brachydactyly[20-22], suggesting a role for ATP1B1 in these disorders.
Deletions within SRO-I (chr1:172,460,683-172,281,412) show high probability to include a disease-locus for ID, microcephaly, and, as previously defined by Lefroy[2], skeletal anomalies including short hands and feet and brachydactyly [Figures 5 and 6A].
Specifically, the SRO-I includes Dynamin-3 (DNM3, OMIM *611445), a gene harboring a 7.9-kb antisense transcript for miR199-214 genes[23]. These two miRs are involved in vertebrate skeletogenesis[24,25], thus suggesting a role for the skeletal phenotype in 1q24 deleted patients[2,6]. Indeed, the phenotype of Cases 2-4 with 1q24q25 deletions fully including the DNM3, with its two guests miR199-214[Figure 6D], share significant pre- and postnatal growth deficiency, microcephaly, and small hand and feet with fifth finger clino-brachydactyly [Figures 1A and B and 2A and B, Table 1], as reported in all patients with a deletion including entirely DNM3[1-2,4,6] [Figures 5B-D and 6A].
In agreement with our data, our probability distribution profiles indicate that deletions for the proximal portion of SRO-D (chr1:177,238,295-178,548,677), indicated as No. 3 (SRO-3, Table 2), are significantly associated with microcephaly, ID, and short hands/feet [Figures 5A-C and 6A]; less with brachydactyly [Figures 5B and 6A], micro/retrognathia and hypertelorism; and not associated at all with kidney abnormalities, hypotonia, speech and walk delay, and dysplastic ears [Supplementary Figures 4 and 5].
BRINP2/FAM5B is the only gene of the region that is intolerant to haploinsufficiency but its role is unknown [Figure 6E and Supplementary Table 4].
In contrast, our probability distribution profiles indicate that deletions for the distal portion of SRO-D are significantly associated only with ID and brachydactyly [Figures 5A and 6A], although it should be noted that the microcephaly area of probability does not encompass this region due to the dubious effect of the deletion in our Patient 5. This region includes CEP350, RALGPS2, TDRD5, and XPR1 genes that are intolerant to haploinsufficiency; all have a low brain expression but none of them is thus far recognized as a disease gene [Supplementary Table 4]. In addition, LIM homeobox 4 (LHX4, OMIM 602146), a gene implicated in the etiology of congenital hypopituitarism[26] (OMIM #262700), was previously evoked as possible candidate gene for growth deficiency[7,27].
Altogether, we have to assume that deletion for the proximal region 1q23.3q24.1 (SRO-P[1]) is associated with kidney anomalies of high penetrance for total PBX1 loss [Supplementary Figures 4 and 5] and ID but not microcephaly [Figure 5]. Microcephaly is fully associated with deletions of more than one SROs (SRO-2, -I, and -D, Figures 6 and 7) and the most favorable new candidate gene is ATP1B1 in SRO-2.
Cases 5 and 6 are puzzling for the apparently different phenotypes in the presence of identical deletion. Indeed, Case 5 is a healthy adult and Case 6 is still a newborn with a nuanced disorder and perhaps would have been considered healthy if we had not incidentally identified the deletion in the father. Among genes mapping in their 5.9-Mb deleted region, between 172,667,560 and 178,548,677 [Supplementary Table 5], at least two are associated with systemic diseases: TNFSF4 (MIM *603594), related to systemic lupus erythematosus (OMIM#152700), and DARS2 (MIM*610956), involved in recessive leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (MIM#611115). None of these conditions were consistent with the clinical presentation of our Cases 5 and 6. We reasoned that the deletion might have unmasked a maternally inherited recessive variant in that region. However, WES analysis on the mother did not support this hypothesis, suggesting that other genetic or environmental factors may modulate the phenotype associated with this deletion. Indeed, the microcephaly, which is the main feature, observed in our Case 6, is a neurological sign that may be caused by a multitude of disease-causing genes with recessive or dominant inheritance[28]. However, by WES, we did not highlight any possible pathogenic variants in all genes associated with microcephaly having a frequency < 1%[28] that could explain this trait in our Case 6.
Similarly, IUGR, which characterized the prenatal life of our Case 6, is an end result of various etiologies that include maternal, placental, fetal, and genetic factors[29]. Looking at the clinical history of our family [Figure 3], IUGR was documented not only in the child carrying the 1q deletion, but also in her brother without this deletion (Subject III.2), and even in the IUFD at 39th week of gestation (Subject III.3). Indeed, by WES data, we identified in the mother’s genome, two heterozygous missense variants, c.796C>T:p.R266W and c.1279G>A:p.A427T in TXBAS1[Supplementary Figure 3], a gene having a possible role in thrombotic events[30,31]. Since both variants are rare (AF< 0.001%), predicted deleterious in several databases, and almost certainly in trans, only one being present in the first son of the couple, the two variants most likely represent a risk factor for the recurrent pregnancy losses and IUGR observed in this family.
Taken together, the link between 1q deletion identified in this family and the phenotype in our patient remains elusive. A long-term clinical follow-up of our newborn patient will help to clarify whether this deletion represents a benign CNV or a rearrangement showing incomplete penetrance.
In conclusion, we confirmed and identified several genes whose haploinsufficiency appears crucial in the manifestation of the main phenotypic abnormalities associated with 1q23.3q25.2 deletions [Supplementary Table 6]. In particular, PBX1, in addition to its well-known role in kidney abnormalities, is strongly associated with ID and contributes to the behavioral traits along with psychiatric disorders. DNM3 and LHX4 are hereby confirmed as responsible for growth retardation[7,27] while ATP1B1 represents a new candidate gene for microcephaly.
It should however be underlined that, apart from SRO-1, the other three SROs contain genes belonging to different TADS, some of which are interrupted by the deletion (http://3dgenome.org).
We cannot therefore rule out that some phenotypic abnormalities are due to an altered expression of some of the non-deleted genes following the breakdown of the TADs, rather than the haploinsufficiency of specific genes[32].
Finally, we propose a method to computationally predict the probability that a given DL lies in a specific genomic segment. Although this approach may be hampered by long-term position effects of regulatory elements, synergistic cooperation of several genes, and incomplete clinical assessment, it can be useful, especially for contiguous gene syndromes that show a complex pattern of clinical characteristics. Obviously, functional approaches are needed to warrant its reliability.
Declarations
AcknowledgmentsWe would like to gratefully acknowledge the family participating in this study.
We thank Dr. Lucialba Rapisarda, expert in modeling and simulation of complex systems, for her useful advice in preparing probability related graphs.
Authors’ contributionsMade substantial contributions to conception and design of the study, performed data analysis and interpretation, wrote the original draft: Fichera M, Bonaglia MC
Investigation: Saccuzzo L, Bertuzzo S, Cavallini A, Bonaglia MC
Provided clinical data: Marelli S, Cavallini A, Romaniello R, Kocova M, Citterio A, Fanizza I, Trabacca A, Pagliazzi A, Guarducci S, Giglio S
Writing - Review & Editing: Fichera M, Zuffardi O, Bonaglia MC
Supervision and acquisition of the financial support for the project leading to this publication: Bonaglia MC
Availability of data and materialsData supporting our findings are published as supplementary information in the journal.
The software is available on request.
Financial support and sponsorshipThis work was supported by the Italian Ministry of Health (2758027) to Bonaglia MC.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethics approval and consent to participateThe study was conducted in accordance with the Declaration of Helsinki and national guidelines. Written informed consent was obtained from each subject’s parent(s) or their guardian, and the study was approved by E. Medea Scientific Institute Research Ethics Committee approval (Prot. No. 007/15-CE).
Consent for publicationPatient photographs are shown in cases where parents have consented to publication.
Copyright© The Author(s) 2020.
REFERENCES
1. Chatron N, Haddad V, Andrieux J, Desir J, Boute O, et al. Refinement of genotype-phenotype correlation in 18 patients carrying a 1q24q25 deletion. Am J Med Genet A 2015;167:1008-17.
2. Lefroy H, Fox O, Javaid MK, Makaya T, Shears DJ. 1q24 deletion syndrome. Two cases and new insights into genotype-phenotype correlations. Am J Med Genet A 2018;176:2004-8.
3. Filges I, Bischof-Renner A, Röthlisberger B, Potthoff C, Glanzmann R, et al. Panhypopituitarism presenting as life-threatening heart failure caused by an inherited microdeletion in 1q25 including LHX4. Pediatrics 2012;129:e529-34.
4. Burkardt DD, Rosenfeld JA, Helgeson ML, Angle B, Banks V, et al. Distinctive phenotype in 9 patients with deletion of chromosome 1q24-q25. Am J Med Genet A 2011;155:1336-51.
5. Della Monica M, Lonardo F, Faravelli F, Pierluigi M, Luquetti DV, et al. A case of autism with an interstitial 1q deletion (1q23.3-24.2) and a de novo translocation of chromosomes 1q and 5q. Am J Med Genet A 2007;143:2733-7.
6. Ashraf T, Collinson MN, Fairhurst J, Wang R, Wilson LC, et al. Two further patients with the 1q24 deletion syndrome expand the phenotype: A possible role for the miR199-214 cluster in the skeletal features of the condition. Am J Med Genet A 2015;167:3153-60.
7. Lam F, Morris C. Nine year old boy with chromosome 1q23.3-q25.1 deletion. Am J Med Genet A 2016;170:3013-7.
8. Hoshina T, Seto T, Shimono T, Sakamoto H, Okuyama T, et al. Narrowing down the region responsible for 1q23.3q24.1 microdeletion by identifying the smallest deletion. Hum Genome Var 2019;6:47.
9. Sun M, Lou J, Li Q, Chen J, Li Y, et al. Prenatal findings and molecular cytogenetic analyses of a
10. Arcaleni E. Secular trend and regional differences in the stature of Italians. J Anthropol Sci 2012;90:233-7.
11. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1-33.
12. Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 2015;10:1556-66.
13. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297-303.
14. Chase MB, Baek SJ, Purtell DC, Schwartz S, Shen RF. Mapping of the human thromboxane synthase gene (TBXAS1) to chromosome 7q34-q35 by two-color fluorescence in situ hybridization. Genomics 1993;16:771-3.
15. Slavotinek A, Risolino M, Losa M, Cho MT, Monaghan KG, et al.
16. Le Tanno P, Breton J, Bidart M, Satre V, Harbuz R, et al.
17. Genomic Relationships, Novel Loci. Pleiotropic mechanisms across eight psychiatric disorders. Cell 2019;179:1469-82.
18. Clausen MV, Hilbers F, Poulsen H. The structure and function of the Na,K-ATPase isoforms in health and disease. Front Physiol 2017;8:371.
19. Lanciotti A, Brignone MS, Molinari P, Visentin S, De Nuccio C, et al. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the
20. Krakow D, Vriens J, Camacho N, Luong P, Deixler H, et al. Mutations in the gene encoding the calciumpermeable ion channel TRPV4 produce spondylometaphyseal dysplasia, kozlowski type and metatropic dysplasia. American Journal of Human Genetics 2019;84:307-15.
22. Thibodeau ML, Peters CH, Townsend KN, Shen Y, Hendson G, et al. Compound heterozygous TRPV4 mutations in two siblings with a complex phenotype including severe intellectual disability and neuropathy. Am J Med Genet A 2017;173:3087-92.
23. Watanabe T, Sato T, Amano T, Kawamura Y, Kawamura N, et al. Dnm3os, a non-coding RNA, is required for normal growth and skeletal development in mice. Dev Dyn 2008;237:3738-48.
24. Lin EA, Kong L, Bai XH, Luan Y, Liu CJ. miR-199a, a bone morphogenic protein 2-responsive MicroRNA, regulates chondrogenesis via direct targeting to Smad1. J Biol Chem 2009;284:11326-35.
25. Shi K, Lu J, Zhao Y, Wang L, Li J, et al. MicroRNA-214 suppresses osteogenic differentiation of C2C12 myoblast cells by targeting Osterix. Bone 2013;55:487-94.
26. Xatzipsalti M, Voutetakis A, Stamoyannou L, Chrousos GP, Kanaka-Gantenbein C. Congenital hypopituitarism: various Genes, various phenotypes. Horm Metab Res 2019;51:81-90.
27. Nishimura A, Hiraki Y, Shimoda H, Nishimura G, Tadaki H, et al. De novo deletion of 1q24.3-q31.2 in a patient with severe growth retardation. Am J Med Genet A 2010;152:1322-5.
28. Boonsawat P, Joset P, Steindl K, Oneda B, Gogoll L, et al. Elucidation of the phenotypic spectrum and genetic landscape in primary and secondary microcephaly. Genet Med 2019;21:2043-58.
29. Sharma D, Shastri S, Farahbakhsh N, Sharma P. Intrauterine growth restriction-part 1. J Matern Fetal Neonatal Med 2016;29:3977-87.
30. Tsujita Y, Kinoshita M, Tanabe T, Iwai N. Role of a genetic variation in the promoter of human thromboxane synthase gene in myocardial infarction. Atherosclerosis 2000;153:261-2.
31. Yi X, Ming B, Wang C, Chen H, Ma C. Variants in COX-2, PTGIS, and TBXAS1 are associated with carotid artery or intracranial arterial stenosis and neurologic deterioration in ischemic stroke patients. J Stroke Cerebrovasc Dis 2017;26:1128-35.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Fichera M, Saccuzzo L, Bertuzzo S, Marelli S, Cavallini A, Romaniello R, Kocova M, Citterio A, Fanizza I, Trabacca A, Pagliazzi A, Guarducci S, Giglio S, Zuffardi O, Bonaglia MC. Assigning single clinical features to their disease-locus in large deletions: the example of chromosome 1q23-25 deletion syndrome. J Transl Genet Genom 2020;4:114-32. http://dx.doi.org/10.20517/jtgg.2020.16
AMA Style
Fichera M, Saccuzzo L, Bertuzzo S, Marelli S, Cavallini A, Romaniello R, Kocova M, Citterio A, Fanizza I, Trabacca A, Pagliazzi A, Guarducci S, Giglio S, Zuffardi O, Bonaglia MC. Assigning single clinical features to their disease-locus in large deletions: the example of chromosome 1q23-25 deletion syndrome. Journal of Translational Genetics and Genomics. 2020; 4(3): 114-32. http://dx.doi.org/10.20517/jtgg.2020.16
Chicago/Turabian Style
Fichera, Marco, Lucia Saccuzzo, Sara Bertuzzo, Susan Marelli, Anna Cavallini, Romina Romaniello, Mirjana Kocova, Andrea Citterio, Isabella Fanizza, Antonio Trabacca, Angelica Pagliazzi, Silvia Guarducci, Sabrina Giglio, Orsetta Zuffardi, Maria Clara Bonaglia. 2020. "Assigning single clinical features to their disease-locus in large deletions: the example of chromosome 1q23-25 deletion syndrome" Journal of Translational Genetics and Genomics. 4, no.3: 114-32. http://dx.doi.org/10.20517/jtgg.2020.16
ACS Style
Fichera, M.; Saccuzzo L.; Bertuzzo S.; Marelli S.; Cavallini A.; Romaniello R.; Kocova M.; Citterio A.; Fanizza I.; Trabacca A.; Pagliazzi A.; Guarducci S.; Giglio S.; Zuffardi O.; Bonaglia MC. Assigning single clinical features to their disease-locus in large deletions: the example of chromosome 1q23-25 deletion syndrome. J. Transl. Genet. Genom. 2020, 4, 114-32. http://dx.doi.org/10.20517/jtgg.2020.16
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 12 clicks
Cite This Article 12 clicks

 Like This Article 31
likes
Like This Article 31
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.