
EHMT1 pathogenic variants and 9q34.3 microdeletions share altered DNA methylation patterns in patients with Kleefstra syndrome

Abstract
Aim: Kleefstra syndrome (KS) is a rare neurodevelopmental disorder caused by haploinsufficiency of the euchromatic histone lysine methyltransferase 1 gene, EHMT1, due to either a submicroscopic 9q34.3 deletion or a pathogenic EHMT1 variant. KS is characterized by intellectual disability, autistic-like features, heart defects, hypotonia and distinctive facial features. Here, we aimed to (1) identify a unique DNA methylation signature in patients with KS, and (2) demonstrate the efficacy of DNA methylation in predicting the pathogenicity of copy number and sequence variants.
Methods: We assayed genome-wide DNA methylation at > 850,000 CpG sites in the blood of KS patients (n = 10) carrying pathogenic variants in EHMT1 or 9q34.3 deletions, as compared to neurotypical controls (n = 42). Differentially methylated sites were validated using additional KS patients (n = 10) and controls (n = 29) to assess specificity and sensitivity of these patterns.
Results: The DNA methylation signature of KS demonstrated high sensitivity and specificity; controls and KS patients with a confirmed molecular diagnosis were classified correctly. In additional individuals with EHMT1 alterations, including frameshift or missense variants and partial gene duplications, DNA methylation classifications were consistent with clinical presentation. Furthermore, genes containing differentially methylated CpG sites were enriched for functions related to KS features, including heart formation and synaptic activity.
Conclusion: The KS DNA methylation signature did not differ in patients with deletions and variants, supporting haploinsufficiency of EHMT1 as the likely causative mechanism. Beyond this finding, it provides new insights into epigenetic dysregulation associated with KS and can be used to classify individuals with uncertain genomic findings or ambiguous clinical presentations.
Keywords
Introduction
Epigenetic regulators, including chromatin remodelers and enzymes that write, read or erase epigenetic marks, are essential to healthy human development[1,2]. Chromatin states and epigenetic patterns play a key role in regulating transcriptional profiles specific to cellular identity and developmental timing[3,4]. This is especially important during neurodevelopment for which developmental processes necessitate a complex orchestration of gene expression and environmental signals[5,6]. Recent research into the role of epigenetic regulators in human disease has demonstrated that genes encoding this machinery, termed “epigenes”, are linked to Mendelian neurodevelopmental disorders (NDDs)[2,7]. To date, approximately 70 epigenes have been implicated in 82 distinct conditions, many of which are characterized by intellectual disability (ID) and growth dysregulation[8,9].
At the molecular level, our group and others have found that these NDDs are characterized by aberrant DNA methylation (DNAm) patterns[10,11]. DNAm refers to the addition of a methyl group to cytosine, typically in the context of a cytosine-guanine dinucleotide or CpG. CpG dense regions, known as CpG islands, are found at 70% of gene promoters; in this context, DNAm is usually a repressive mark, corresponding to the silencing of gene activity[12]. DNAm found in enhancers, gene bodies and intergenic regions have a more complex relationship with gene activity and may associate with the repression or activation of genes. Importantly, there is cross-talk between epigenetic marks, such as DNAm and histone 3 lysine 9 (H3K9) trimethylation, exhibiting spatial and temporal co-localization[13,14]. As such, mutations in epigenes encoding enzymes involved in regulation of histone modifications and chromatin packaging often result in downstream DNA methylation alterations in patients carrying these mutations[10,11,15-17]. These altered DNAm patterns reflect genome-wide transcriptional changes at the downstream target genes that constitute the molecular underpinnings of the pathophysiology of the associated NDD. As such, we hypothesize that through a similar cross-talk mechanism between DNAm and H3K9 methylation, pathogenic variants in EHMT1 can affect DNAm patterns
Kleefstra syndrome or KS (MIM 610253) is caused by haploinsufficiency of the euchromatic histone lysine methyltransferase 1 gene, EHMT1[18,19]. EHMT1 encodes a histone methyltransferase that catalyzes mono- and dimethylation of H3K9. As an important transcriptional repressor, EHMT1 is expressed in most human tissues and is overexpressed in a variety of human cancers[20]. KS is caused by either a heterozygous 9q subtelomeric deletion, which overlaps with part or all of EHMT1 (50%) or a heterozygous pathogenic variant in EHMT1, including frameshift, missense and nonsense mutations (50%)[21,22]. Features of KS include moderate to severe ID, childhood hypotonia, seizures, heart defects and characteristic facial features including brachy(-micro)cephaly, synophrys, cupid bowed upper lip and prominent jaw[18,23-25]. As well, urogenital and renal complications, psychiatric disorders, and features of autism spectrum disorder (ASD) are often present. Males and females are affected equally[22]. There is some evidence of genotype-phentoype correlation in that individuals with EHMT1 pathogenic variants and those with a small 9q34.3 deletion (< 1 Mb) have similar clinical findings, whereas individuals with larger deletions (> 1 Mb) can have more severe ID and more medical problems[19,21,26].
Here, we report the differential DNAm patterns associated with KS. We use these patterns to derive a predictive model to classify individuals with EHMT1 sequence variants of uncertain significance or ambiguous clinical presentations. We found that patients with KS caused by either 9q34.3 microdeletions or pathogenic EHMT1 variants exhibit a specific DNAm signature that is unique from both typically developing controls and individuals with similar epigene-related disorders. As well, these differentially methylated CpG sites map to genes enriched for heart and brain development.
Methods
Research participants
Informed consent was obtained from all research participants according to the protocol approved by the Research Ethics Board of the Hospital for Sick Children (REB# 1000038847). Individuals were recruited through the Division of Clinical and Metabolic Genetics at the Hospital for Sick Children, Toronto, Ontario; Department of Medical Genetics, Alberta Children’s Hospital Research Institute, Calgary, Alberta; Baylor College of Medicine, Houston, Texas; Prevention Genetics, Marshfield, Wisconsin; University Hospitals, Cleveland, Ohio; Seoul National University Children’s Hospital, Seoul, Korea; and Department of Pathology and Molecular Medicine, McMaster University, Hamilton, Ontario.
KS patients were divided into groups based on age, diagnosis and molecular findings [Table 1] and availability of clinical phenotype data; importantly, the frequency of deletions and variants in this sample is reflective of the reported frequency in the greater population of KS cases. The first group encompassed individuals over the age of one year with a confirmed clinical diagnosis of KS and a pathogenic EHMT1 sequence variant or 9q34 deletion reported by a molecular diagnostic laboratory. These individuals were separated into discovery and validation groups (n = 10 and n = 5, respectively), such that molecular underpinnings, sex, and age were represented within each group. For all patients in the discovery cohort, clinical and molecular diagnoses were defined by certified clinical and molecular geneticists, respectively. Individuals younger than 1 year represented an additional validation group (n = 5), and individuals with EHMT1 variants but little or no phenotypic information available at the time of data analysis (n = 5) represented an “unknown” test group. Two additional individuals who both carried duplications of chromosome 9q34 were also included in analyses. The presence of specific KS features, such as hypotonia, microcephaly, or heart defects, was limited to a subset of KS patients, and as such, were not included as variables in the statistical analysis. Also, since only one individual with KS carried a deletion greater than 1 Mb, patient KS17_I, deletion size was not considered in the analysis.
Clinical diagnoses and molecular findings of subjects with EHMT1 variants and deletions
| Cohort | ID | Diagnosis | Age (years) | Sex | Molecular | Deletion coordinates (hg38)/ protein change (NM_024757) | Previous publications |
|---|---|---|---|---|---|---|---|
| Discovery | KS1_T | KS | 1 | F | 9q34 deletion | chr9:137728310-137989926 | |
| KS2_T | KS | 1 | F | EHMT1 variant (missense) | p.(P809R) | ||
| KS3_T | KS | 3 | F | 9q34 deletion | chr9:137811023-137967082 | Yatsenko et al.[21] (P37) | |
| KS4_T | KS | 3 | F | EHMT1 variant (frameshift) | p.(V1026Qfs*150) | ||
| KS5_T | KS | 4 | M | 9q34 deletion | NA (~0.4Mb) | Willemsen et al.[26] (P11) | |
| KS6_T | KS | 4 | F | 9q34 deletion | chr9:137620211-137944399 | Yatsenko et al.[21] (P44) | |
| KS7_T | KS | 6 | M | 9q34 deletion | NA (0.32 Mb) | ||
| KS8_T | KS | 7 | M | 9q34 deletion | chr9:137620211-137988669 | Yatsenko et al.[21] (P43) | |
| KS9_T | KS | 8 | M | 9q34 deletion | chr9:137294642-137987222 | ||
| KS10_T | KS | 25 | F | EHMT1 variant (nonsense) | p.(R246*) | ||
| Validation (≥ 1 year) | KS11_V | KS | 1 | M | 9q34 deletion | chr9:137599274-137709523 | |
| KS12_V | KS | 1 | M | 9q34 deletion | chr9:137801987-137862641 | ||
| KS13_V | KS | 5 | M | EHMT1 variant (nonsense) | p.(R260*) | Kleefstra et al.[19] (P19) | |
| KS14_V | KS | 7 | F | 9q34 deletion and 4p duplication | chr9:137586180-138197466 | Willemsen et al.[26] (P12) | |
| KS15_V | KS | 25 | F | 9q34 deletion | NA (subtelomeric) | Willemsen et al.[26] (P10) | |
| Validation (< 1 year) | KS16_I | KS | 0.75 | M | 9q34 deletion | chr9:137513779-138231664 | |
| KS17_I | KS | 0.01 | F | 9q34 deletion | chr9:136914736-138114821 | ||
| KS18_I | KS | 0.1 | F | 9q34 deletion | chr9:137484248-137989926 | ||
| KS19_I | KS | 0.02 | F | 9q34 deletion | chr9:137286411-138125937 | ||
| KS20_I | KS | 0.8 | F | 9q34 deletion | NA (~3.2Mb) | Yatsenko et al.[21] (P14) | |
| Unknown | U1 | ASD | NA | M | EHMT1 variant (frameshift) | p.(E181Gfs*5) | |
| U2 | likely KS | 21 | F | EHMT1 variant (frameshift) | p.(A643Pfs*9) | ||
| U3 | ASD | NA | M | EHMT1 variant (missense) | p.(V402L) | ||
| U4 | ASD | NA | M | EHMT1 variant (missense) | p.(F613Y) | ||
| U5 | ASD | NA | F | EHMT1 variant (missense) | p.(L724P) | ||
| Partial EHMT1 duplication | Dup1 | unknown | 1 | M | 9q34 duplication | chr9:137819943-137988669 | |
| Dup2 | unknown | 17 | M | 9q34 duplication | chr9:137819954-137871875 |
Banked DNA samples from age- and sex-matched neurotypical participants (n = 42) were included as a control group. These individuals were recruited from the Hospital for Sick Children and were deemed typically developing by physician or parental questionnaires. DNA methylation data for individuals with Nicolaides-Baraitser syndrome, an neurodevelopmental disorder (NDD) caused by pathogenic variants in SMARCA2, were downloaded from the Gene Expression Omnibus database, accession number: GSE125367[11]. These individuals were included as an additional “control” group to assess the specificity of the DNAm patterns to KS, as opposed to Nicolaides-Baraitser syndrome, an NDD with similar features, which is also caused by epigenetic dysregulation.
DNAm data generation and preprocessing
Genomic DNA was extracted from peripheral blood samples and bisulfite converted using the protocols described in Chater-Diehl et al.[11]. Converted DNA was then assayed for DNAm levels on the Illumina Infinium MethylationEPIC array (EPIC array; > 850,000 CpG sites) at The Center for Applied Genomics (TCAG), Hospital for Sick Children Research Institute, Toronto, Ontario, Canada in accordance with the manufacturer’s protocols. Samples were randomly stratified across chips and run in two batches but balanced for case/control proportions and sex.
Raw data were then processed in R statistical software, using the package minfi[27]. Quality control measures included removing probes that failed detection P-value, meaning the signal was not significantly above background noise, as well as probes mapping to X and Y chromosomes, cross-reactive probes and SNP probes[28,29]. All criteria and methods for pre-processing are fully described in Chater-Diehl et al.[11]. Following these steps, data underwent background signal subtraction and control normalization also using minfi[29]. The normalized data consisted of 774,583 methylation sites or CpGs for each sample. DNAm, measured in β values, ranges 0-1 representing percent methylation.
DNAm signature derivation
Prior to statistical analysis, underlying proportions of monocytes, neutrophils, CD4T, CD8T, natural killer cells and B cells were estimated from the DNAm data using the Houseman algorithm[30]. At each CpG site, a two-group comparison of KS discovery cases vs. controls was performed using limma regression, accounting for sex, age, batch and estimated blood cell proportion covariates[31]. CpG sites found to be differentially methylated between cases and controls were reported if they met both a statistical significance [false discovery rate (FDR)-corrected P-value < 0.01] and a minimum effect size (absolute Δβ >10%). Δβ represents the difference in average DNAm (β) between groups. Principal component analysis (PCA) and hierarchical clustering were generated using Qlucore Omics Explorer (QOE, www.qlucore.com).
SVM model classification
Statistically significant CpG sites, i.e., the DNAm signature, were used as input into a machine-learning algorithm, support vector machine (SVM), to generate a predictive classification model. To remove noise and to filter out information that did not improve the efficacy of the model, we first removed redundant sites. Any methylation site that was highly correlated (r > 0.9) with any other site was removed, leaving 429 CpG sites. We then built an SVM model using the R package caret (for details of model training and validation, see Butcher et al.[10])[32]. The classification model generated by SVM was then applied to all remaining samples. The output of this model was a probability score indicating likelihood of having KS or a genomic alteration that causes KS.
GO analysis
Gene ontology (GO) enrichment analysis was performed on the KS signature sites using GREAT (Genomic Regions Enrichment of Annotations Tool)[33]. We used a custom “background” that included all 774,583 CpG sites that passed quality control. “Basal+extension” was used to identify associated genes, using the following modified parameters: constitutive 5.0 kb upstream and 1.0 kb downstream, up to 10.0 kb maximum extension. We also refined the output by requiring that significant terms contain two or more gene hits.
Results
Identifying a DNA methylation signature for Kleefstra syndrome
To define a DNAm signature associated with KS, DNA from KS patients and neurotypical controls was extracted from blood and assayed using the EPIC array, generating high-quality measurements at 774,583 CpG sites. Ten unrelated individuals with a confirmed clinical diagnosis of KS, samples KS1_T - KS10_T, and pathogenic variants in EHMT1 or microdeletions of 9q34.3, which included partial or full deletions of EHMT1 (n = 3 and n = 7, respectively; n = 6 females; age 1-25 years) were compared to 42 neurotypical controls (n = 21 females; age 1-28 years). Since we combined data from patients with pathogenic variants in EHMT1 and those with 9q34.3 microdeletions together, our analyses identified DNAm changes common to both molecular causes (analysis of this cohort stratified by molecular alteration did not identify differential methylation; see below). Figure 1 shows the genomic position of all deletions and variants, converted to hg38 genome build. For patients KS5_T and KS7_T, deletion coordinates were not available. KS5_T carried a 0.4-Mb deletion and KS7_T carried a 0.32-Mb deletion, both of which overlapped EHTM1.
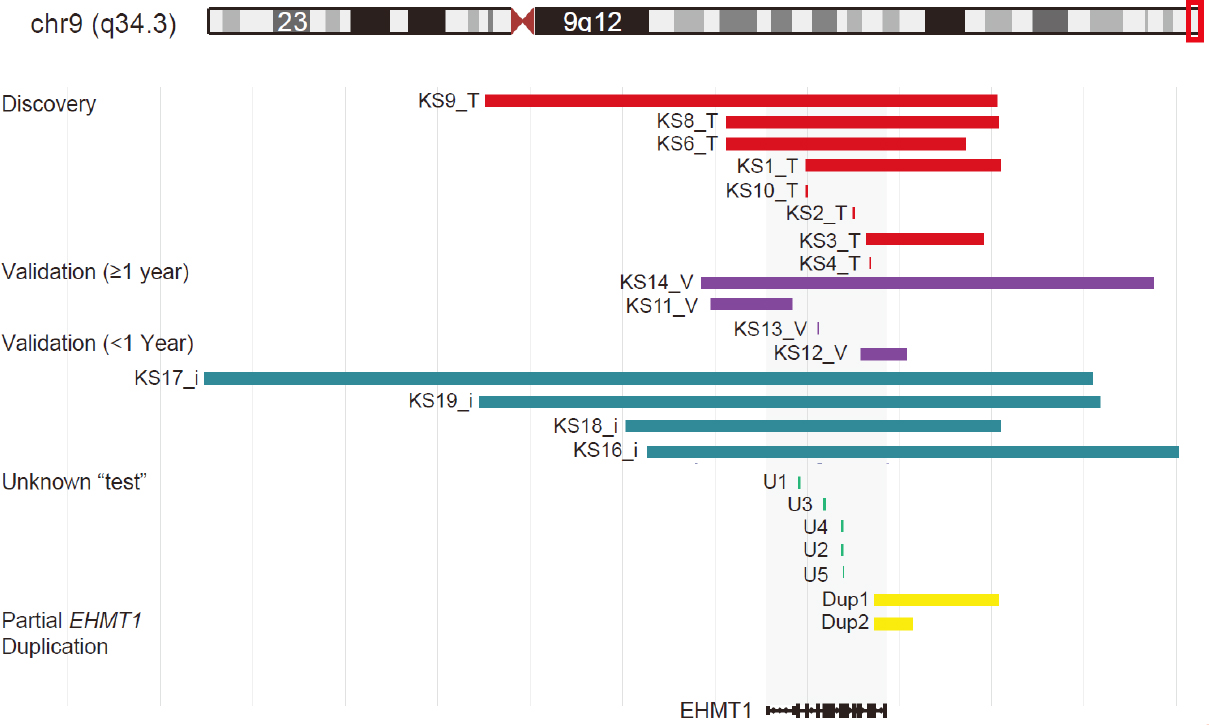
Figure 1. Mapping of gene variants and microdeletions to chromosome 9q34.3. Schematic of all variants relative to EHMT1 (hg38). Variants are colored by analysis group: discovery (n = 8; 2 not depicted), validation older than 1 year (n = 4; 1 not depicted), validation samples younger than 1 year (n = 4; 1 not depicted), unknown test samples with no phenotype information and partial EHMT1 duplication (n = 5 and n = 2, respectively). Genomic coordinates were not available for KS15_T, KS15_T, KS15_V, and KS20_I
Differentially methylated CpG sites were identified using a linear regression which included sex, age, blood cell proportions and array batch as covariates. There was no significant difference in estimated blood cell proportions between groups (all FDR-corrected P-values > 0.05). A total of 598 CpG sites differed significantly between individuals with KS and controls at an FDR-corrected P-value < 0.01 and a minimum mean difference between groups (Δβ) of 10% [Supplementary Table 1]. Also, no CpG site was significantly associated with age (FDR P-value > 0.05). The 598 CpG sites constituted a “DNAm signature” of KS. Both hierarchical clustering and principal component analysis of DNAm values at the signature sites clearly distinguished the KS case group from neurotypical controls [Figure 2]. Of note, 484 signature sites (81%) were hypomethylated in KS patients as compared to controls. Furthermore, CpG sites mapping to CpG islands, commonly found at gene promoters, were significantly underrepresented as compared to all CpG sites that passed quality control [Supplementary Figure 1].
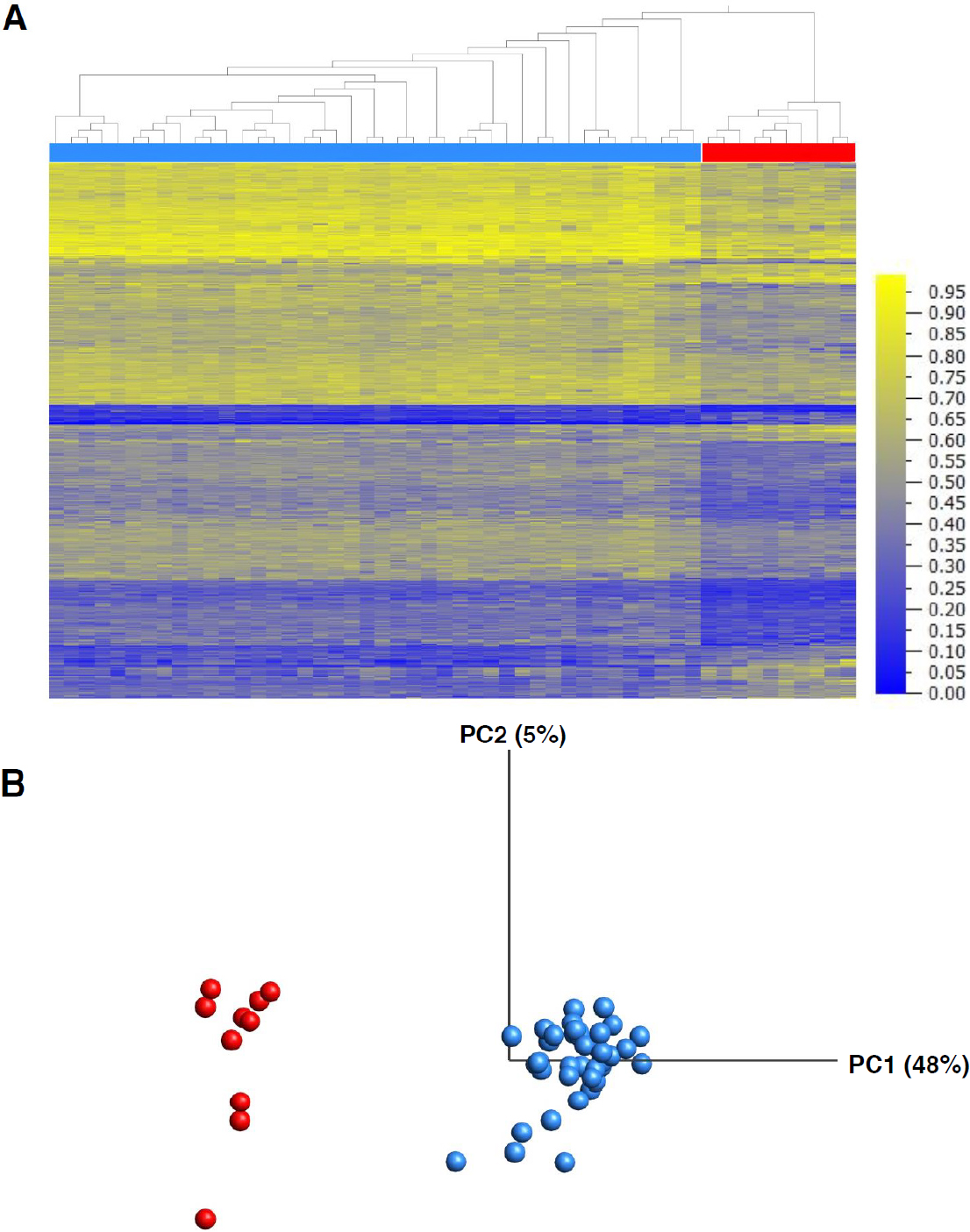
Figure 2. Clustering of KS patients (n = 10) and controls (n = 42) at 598 KS signature sites. Heatmap with samples ordered by Euclidean clustering (A); and principal component analysis illustrating differential methylation signals between KS discovery group and controls (B). Heatmap colors represent percent methylation (b)
Ontology of KS signature genes
A number of genes containing multiple differentially methylated CpGs sites or “signature sites” functioned in pathways related to the KS phenotype. These included: ELAVL4 (4 promoter CpGs; mean Δβ = -11.4%), involved in neuron-specific RNA processing; glutamate receptor GRIA1 (2 promoter CpGs; mean Δβ = -11.4%); CHRND1 (2 promoter CpGs; mean Δβ = -11.4%), a muscle acetylcholine receptor subunit, involved in neuromuscular transmission; and PDE4D. Pathogenic variants in the latter are associated with acrodysostosis 2, a skeletal disorder characterized by facial anomalies and ID[34]. PDE4D contained two hypomethylation CpGs approximately 110 bp apart and located within 1.5 kb upstream of the transcription start site, cg18804667 (Δβ = -16.1%) and cg00322656 (Δβ = -10.7%). As such, we performed gene ontology (GO) analysis on the 598 signature sites using GREAT[33]. GREAT identified 130 genes proximal to the signature sites, which met the criteria of less than 15 kb upstream or 11 kb downstream of the gene’s transcription start site, revealing that the majority of signature sites (408) were intergenic. Enriched terms or pathways in cellular components and human phenotype ontology are listed in Table 2. The most prominent finding was the identification of pathways and processes involved in neuronal and synaptic function across all ontologies.
Top ranking GO terms for genes mapping to KS signature sites
| Ontology | Term name | Hyper raw
P-value | Hyper FDR
Q-value | Hyper fold enrichment | CpG hits | Total CpGs | Gene hits | Total genes annotated |
|---|---|---|---|---|---|---|---|---|
| GO cellular component | Postsynaptic membrane | 2.46E-10 | 2.12E-07 | 5.82 | 21 | 4,685 | 8 | 224 |
| Synaptic membrane | 6.69E-09 | 2.89E-06 | 4.59 | 22 | 6,218 | 9 | 294 | |
| Presynaptic membrane | 7.97E-09 | 2.75E-06 | 9.66 | 12 | 1,612 | 5 | 71 | |
| Receptor complex | 1.45E-06 | 4.18E-04 | 3.75 | 19 | 6,582 | 6 | 322 | |
| Postsynapse | 4.00E-06 | 9.87E-04 | 3.13 | 22 | 9,125 | 9 | 422 | |
| Chloride channel complex | 8.60E-05 | 1.86E-02 | 8.59 | 6 | 906 | 2 | 47 | |
| Human phenotype | Hypoplastic heart | 5.17E-06 | 1.72E-02 | 20.86 | 5 | 311 | 2 | 11 |
The single enriched human phenotype was hypoplastic heart. Congenital heart defects are a core feature of KS, including one reported severe presentation of KS with hypoplastic left heart syndrome[35]. Two genes, CHRND and DTNA, containing differentially methylated CpG sites were annotated to this GO term.
Independent validation of Kleefstra syndrome DNA methylation signature
Using the KS DNAm signature, we developed a machine learning classification model capable of categorizing individuals as positive or negative for KS on the basis of their DNAm levels at signature sites. We trained a SVM model on data from the KS discovery group (n = 10) and neurotypical controls (n = 42) used to derive the signature. First, we classified a validation cohort of five unrelated individuals with a clinical KS diagnosis, KS11_V - KS15_V (EHMT1 nonsense variant n = 1 and 9q34.3 deletions n = 4). The SVM model classified all five KS individuals correctly, i.e., as positive for KS, demonstrating high sensitivity of the signature [Figure 3]. Furthermore, we classified an additional validation cohort of five unrelated KS individuals under the age of 1 year, KS16_I - KS20_I; their ages ranged from 2 days to 9 months. Despite training the classification model only on individuals over 1 year old, the model correctly classified all KS infants as positive for KS [Figure 3].
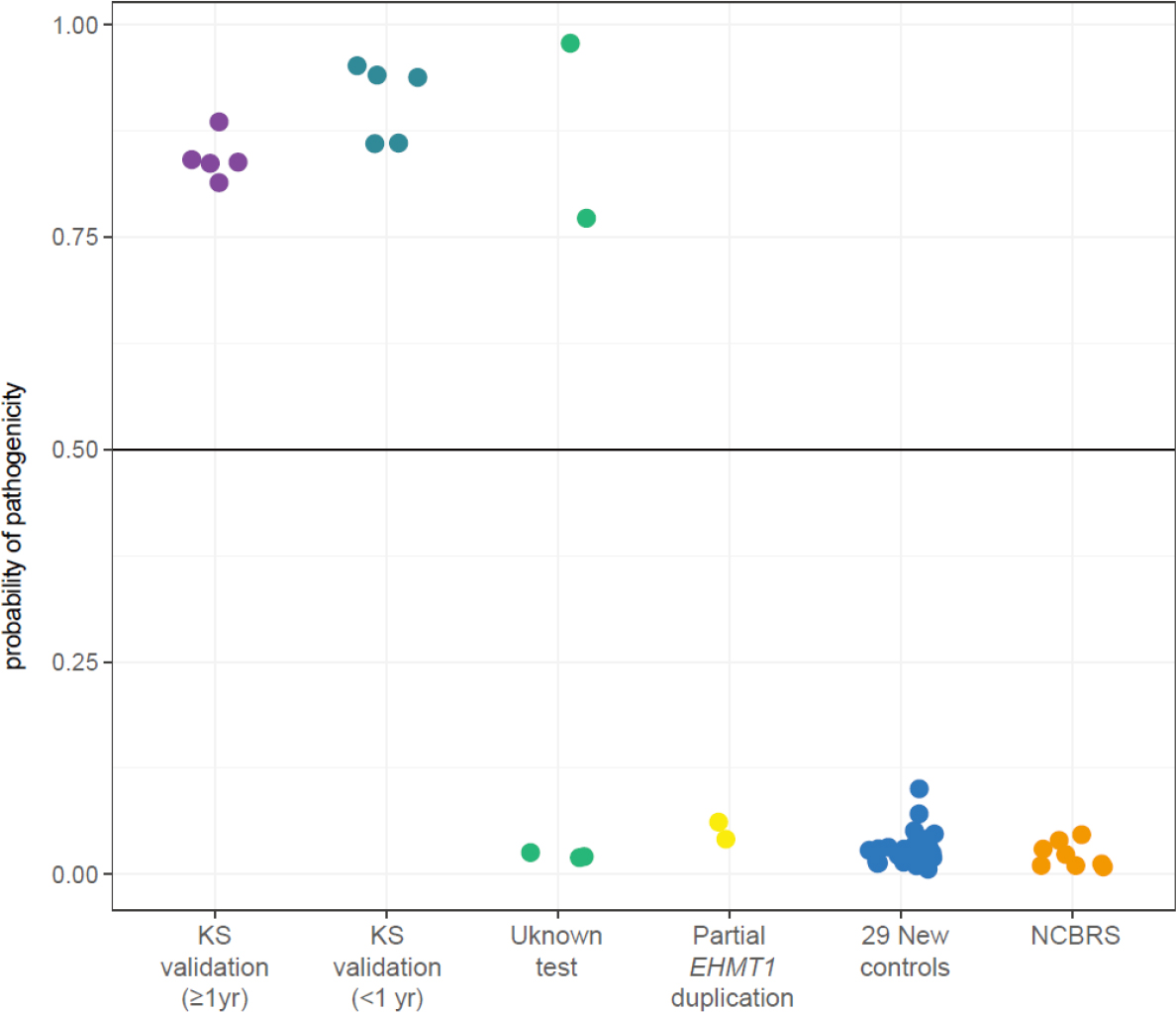
Figure 3. Classification of additional samples with EHMT1 variants or deletions (n = 12) using KS signature. Output of SVM classification model trained on KS signature sites generating the probability of having KS for each sample. Samples classified include 12 individuals in unknown test or validation groups, 29 new controls, and 4 individuals with NCBRS. Horizontal line represents threshold for classifying samples as cases (above line) or controls (below line)
To test the specificity of the KS DNAm signature, we assessed an additional 29 neurotypical controls (n = 14 female, age 1 month to 16 years), all of which classified as negative for KS (i.e., with controls) demonstrating 100% specificity of the signature [Figure 3]. Additionally, we assessed whether the KS DNAm signature could be used to classify patients (n = 8) with Nicolaides-Baraitser syndrome (NCBRS), a neurodevelopmental disorder with some clinical features that overlap KS, including ID, ASD and seizures but with distinct facial characteristics. NCBRS is caused by haploinsufficiency of SMARCA2, which encodes a protein that is part of another epigenetic regulator (SNF/SWI chromatin remodeling complex). Pathogenic variants in SMARCA2 have been shown to be associated with a distinct DNAm signature[11]. All eight samples were classified as negative for KS, with controls (n = 5 female, age 4-15 years), providing further evidence of the specificity of the KS DNAm signature.
Finally, we tested two individuals carrying partial duplications of EHMT1, both of which mapped to the last two exons of the gene[26,27]. Duplications with similar boundaries in individuals with variable phenotypes, including ID and dysmorphic features, have been previously reported as benign[36]. Both patients, Dup1 and Dup2, were classified as negative for KS. Patient Dup1 had dysmorphic features and also carried an unbalanced 31.8-Mb complex rearrangement on chromosome 6p, as well as a microdeletion on 6q27 (0.89 Mb). Patient Dup2 exhibited ID, microcephaly and selective mutism. While a 9q34 duplication has been described in one individual with KS, spanning exons 2-10 of EHMT1, leading to a downstream, premature stop codon and loss of function[37]. DNAm-based classification of these individuals suggests that neither 9q34.3 CNV disrupted EHTM1 gene function that resulted in haploinsufficiency of the EHMT1 protein.
EHMT1 variant classification
Having illustrated the efficacy of the KS DNAm signature in appropriately classifying individuals with clinical diagnosis of KS, we next assessed five individuals with EHMT1 variants for whom we had limited or no phenotypic data available at the time of analysis, samples U1-U5. Following classification, if phenotype information was available (beyond an ASD diagnosis), it was accessed. Four of these individuals, samples U1, U3, U4 and U5, had a diagnosis of ASD and had undergone whole-genome sequencing; the remaining individual, U2, had no phenotypic or clinical information available but carried an EHMT1 variant identified by targeted EHMT1 testing. The variants identified in these five patients were assessed for predicted pathogenicity using Alamut variant annotation software, which applies multiple prediction algorithms [Table 3] and found to be novel in gnomAD (v3).
Predicted pathogenicity of variants in subjects with limited phenotypic information
| Sample | Protein change (NM_024757) | Inheritance | Coding effect | Predicted pathogenicity | ||||
|---|---|---|---|---|---|---|---|---|
| SIFT (score) | PolyPhen-2 (score) | Mutation taster | Align GVGD | DNAm signature classification | ||||
| U1 | p.(E181Gfs*5) | De novo | FS | - | - | - | - | KS |
| U2 | p.(A643Pfs*9) | Unknown | - | - | - | - | KS | |
| U3 | p.(V402L) | Unknown | Missense | Tolerated (0.39) | Benign (0.013) | Polymorphism | C01 | Negative for KS |
| U4 | p.(F613Y) | Unknown | Deleterious (0) | Benign (0.365) | Disease-causing | C0 | Negative for KS | |
| U5 | p.(L724P) | De novo | Tolerated (0.19) | Probably damaging (1.000) | Disease- causing | C0 | Negative for KS | |
Using the KS DNAm signature, the two patients carrying frameshift mutations, U1 and U2, were classified as positive for KS. Following classification, additional clinical information for U1 was obtained and included mild ID, pulmonary stenosis, genital malformations, dysmorphic facial features and mild hypotonia. Such features support a clinical diagnosis of KS. No additional phenotypic information was available for U2. However, given that this individual underwent targeted EHMT1 gene testing, it is likely that her healthcare providers had a high clinical suspicion of KS.
All three individuals with missense variants, U3-U5, were classified as negative for KS [Table 3]. For patient U3, this classification was supported by all pathogenicity prediction algorithms. This individual was reported to have ASD, macrocephaly, and obesity; macrocephaly is not typically reported in KS. For patients U4 and U5, sequence-based pathogenicity predictions were inconsistent. Clinically, patient U4 was described as having ASD, obesity and asthma. Patient U5 was diagnosed with PDD-NOS (pervasive developmental disorder - not otherwise specified), with no history of motor delay or growth abnormalities (50th percentile for height, weight and head circumference at approximately three years). Both phenotypes were inconsistent with KS clinical features, thus supporting non-KS classification.
Comparing DNAm patterns in individuals EHMT1 variants and 9q34.3 microdeletions
Using all individuals that classified positively, we next wanted to assess if DNAm patterns varied by molecular finding. We compared individuals with EHTM1 variants (n = 6) and deletions (n = 16) at all 774,583 CpGs sites and found no CpG sites to be differentially methylated between these two groups of KS individuals (all FDR P-values > 0.05). Furthermore, average methylation changes compared to controls, measured as Δβ, were comparable between individuals with variants and deletions [Figure 4], further supporting the underlying cause of the KS-associated DNAm signature as EHMT1 haploinsufficiency.
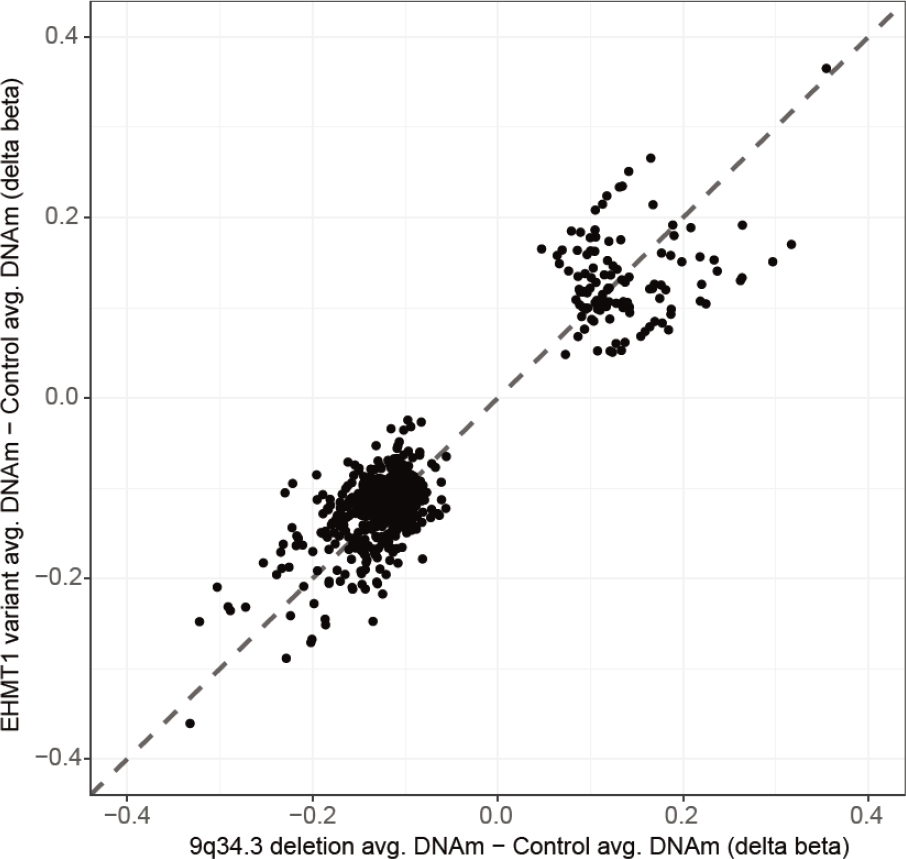
Figure 4. Comparison of mean DNAm changes (Db) in KS patients with variants vs. deletions across 598 KS signature sites. Δβ values calculated in individuals with variants (n = 6) and deletions (n = 16) exhibiting similar magnitudes between groups
Discussion
We identified a genome-wide DNAm signature, associated with haploinsufficiency of the EHMT1 gene product in the peripheral blood of individuals with KS. The signature enabled the classification of both 9q34.3 microdeletions encompassing all or part of EHMT1 and pathogenic EHMT1 variants. Of the 598 signature sites identified, 81% exhibited a loss of methylation in individuals with KS as compared to controls. EHMT1 acts as an H3K9 methyltransferase, typically depositing repressive marks, H3K9me1/2, in euchromatin. However, it has also been shown to methylate non-DNA targets including DNA ligase 1 (LIG1), which once methylated, plays a role in recruiting DNMT1 to hemi-methylated DNA during replication[38]. More specifically, the methylated LIG1 protein more readily binds UHRF1; this binding event recruits UHRF1 to replication sites, binding to hemi-methylated DNA and promoting maintenance of DNA methylation by DNMT1[38]. Furthermore, mouse embryonic stem cells in which Uhrf1 has been knocked down, progressively lose DNAm[39,40]. Therefore, we propose that the loss of DNAm observed in individuals with KS may be a consequence of dysregulated DNA methylation maintenance by DNMT1 due to EHMT1 haploinsufficiency. We also demonstrated that individuals with deletions and variants had indistinguishable DNAm patterns genome-wide. This has previously been reported in other NDDs associated with epigenetic regulators including: pathogenic variants in ARID1B and 6q.25.2 deletions in Coffin-Siris syndrome; pathogenic variants in NSD1 or 5q35.3 deletions in Sotos syndrome; and pathogenic SET1B variants and 12q31.24 deletions in SETD1B-related syndrome[15,16,41].
An important aim of the present study was to generate a broad KS signature that captured the underlying pathophysiological effects of EHMT1 haploinsufficiency on the epigenome, in addition to comparing the impact of CNVs vs. SNVs. To that end, our reported KS signature sites differed somewhat from a DNAm signature of KS recently reported as part of a bioinformatics pipeline of 34 signatures designed to uniquely classify NDDs[42]; this signature was developed using 15 patients with KS, including three individuals with 9q34.3 CNVs, 11 individuals with EHMT1 SNVs, and one individual with a clinical diagnosis of KS but no molecular data. Importantly, the reported signature was constrained to 107 CpGs to reduce “noise” and redundancy, and to optimize output for potential use in diagnostic testing[42]. Of the 107 CpGs sites in this signature, 28 overlapped with the 598 sites in the signature reported here [Supplementary Table 1]. This difference can, in part, be attributed to the platforms used to generate and validate the respective signatures. The signature presented here was generated and validated using only EPIC array data, as compared to Aref-Eshghi et al.[42], who analyzed data from both the EPIC array (assays ~850,000 CpGs) and Infinium HumanMethylation450 array (450K array; assays ~450,000 CpGs; 90% of which are represented on the EPIC array). Therefore all 107 CpG sites in their signature are present on the 450K array[42]. Of our 598 signature sites, approximately half (317) are not represented on the 450K array. Beyond this technical difference, different statistical methods were employed for signature generation. The Aref-Eshghi et al.[42] signature was generated by initially ranking CpG sites on an interaction between P-value and effect size, with no required minimum P-value or multiple testing correction, and the top 1000 CpG sites underwent further analysis, including receiver operating characteristics curve, to select a final set of 100-150 CpGs[42]. Our signature sites were required to meet stringent significance and effect size thresholds (FDR-corrected P-value < 0.01, absolute Δβ > 10%), with no restrictions on signature size. The differences in methods and outcomes highlight two important and overlapping applications of DNAm data, i.e., understanding pathophysiology and use of DNAm data in diagnostics.
GO analysis of the genes identified by our KS signature sites recognized enriched functions and pathways related to KS pathophysiology. Several processes related to neuronal and synaptic function were identified, relevant to the high frequency of ID observed in patients with KS. The top term for GO biological processes was “homophilic cell adhesion via plasma membrane adhesion molecules”. This term was enriched due to signature sites mapping to CHD4, CDH5, and seven γ-protocadherin genes. Protocadherins are neuronal cell surface proteins serving several functions including avoidance of dendritic self-synapsing by conferring self-identity[43]; these genes are crucial for normal synaptic development. Aberrant epigenetic regulation of these genes has also been associated with many NDDs, including Down syndrome and Williams-Beuren syndrome[44,45]. Furthermore, epigenetic dysregulation of protocadherins has been previously implicated in KS pathophysiology; brain tissue of Ehmt1+/- mice display increased H3K9 methylation at protocadherin genes that exhibit dysregulated expression[46]. While both DNA methylation loss and H3K9me2/3 gain support the role of protocadherin dysregulation in KS, we propose that these outcomes may be paradoxically independent, since EHMT1 has been shown to silence transcription by independently acting on both H3K9 and DNAm[47]. In keeping with this model, loci with the greatest changes to H3K9 methylation in Ehmt1+/- mice, showed no consistent DNAm changes[46]. Future studies in human cells are necessary to show if changes to H3K9 and DNAm marks in individuals with KS are indeed uncoupled and result from different EHMT1 functions, as proposed here. Also, such studies would greatly benefit from expression assays in relevant tissue types to directly measure the functional consequences of these dysregulated epigenetic patterns.
In addition to the 10 KS patients in the validation groups, including five infants, who were positively classified by our DNAm signature, we tested an additional seven individuals of interest. Within the validation cases, individuals did not cluster by age [Supplementary Figure 2]; this suggests that the KS signature is not strongly affected by age and likely independent of the dynamic DNAm changes that occur in the first year of life, which occur in part due to normal developmental shifts in blood cell composition[48]. The remaining seven individuals tested all carried genomic variants at 9q34.3: two individuals with partial duplications of EHMT1 and five with single nucleotide variants but limited clinical or no information available. Both patients with duplications were classified as negative for KS, with controls. Such copy number variants are commonly reported as uncertain, as their impact on gene function cannot be ascertained via cytogenetic analysis. On the basis of DNAm classifications, we suggest that the clinical phenotypes of these individuals are likely not related to their EHMT1-asociated CNVs; however, a functional protein assay in these cases would be valuable to confirm our findings.
Patient U1 was one of five individuals with limited clinical or no information available. This individual had a diagnosis of ASD and had undergone whole-genome sequencing, which identified a de novoEHMT1 frameshift variant. Following positive classification of this individual, we learned that he exhibited many features of KS, including mild ID, pulmonary stenosis, dysmorphic facial features and mild hypotonia. This finding speaks to the potential of DNAm signatures for clinical translation, i.e., to be used in concert with genome or exome sequencing to enhance interpretability of genome diagnostics.
A considerable strength of previously reported DNAm signatures is their utility in predicting the pathogenicity of variants of uncertain significance (VUS)[10,11,15]. Pathogenic missense variants are rare but present in KS[49], Thus, we sought to assess the power/utility of the KS signature in classifying missense variants. We included one patient in the discovery group used to derive the DNAm signature, who carried a pathogenic missense variant, P809R, and therefore expected the KS DNAm signature to have clinical utility in this regard. Of note, this variant was predicted to be pathogenic/damaging in Polyphen (1.000), Align GVGD (C65), and SIFT (0). We included three individuals with EHMT1 missense variants in the “test” group and found that each was classified as negative for KS, with controls. The clinical information for these individuals, although limited, was consistent with the DNAm signature prediction.
Although Kleefstra syndrome has a clinically recognizable phenotype, affected individuals exhibit a range of cognitive and behavioral characteristics. Currently, there is little understanding of the relationship between genotype and phenotype. The complex genotype-phenotype relationship in KS will require further study using a large KS cohort with well characterized phenotypes. Our work presented here demonstrates that EHMT1 variants and 9q34.3 deletions share a DNAm signature, further supporting the underlying cause of KS as EHMT1 haploinsufficiency. Further epigenetic research in KS has the potential to elucidate the relationship between genotype and phenotype by refining the DNAm signature and identifying DNAm alterations associated with specific features of KS or specific molecular variants. Therefore, building upon this work to identify genes with altered regulation and expression patterns in KS will provide novel insights into the molecular pathophysiology of this disorder.
Declarations
AcknowledgmentsWe would like to thank all of the patients and families for participating in our research studies and the physicians, clinical staff and research staff for their assistance with patient recruitment. We would also like to thank Chunhua Zhang, Youliang Lou and Khadine Wiltshire for their contributions to this work.
Authors’ contributionsAnalyzed and interpreted the data, generated figures/tables, and wrote the manuscript: Goodman SJ
Collected clinical data, enrolled patients, managed collaborations, integrated clinical findings: Cytrynbaum C
Provided DNA samples and clinical data and provided input on study design and manuscript preparation: Chung BHY, Kellam B, Keller M, Ko JM, Caluseriu O, Grafodatskaya D, McCready E, Perrier R, Yeung KS, Ho-Ming L, Machado J, Stavropoulos DJ, Scherer SW, Innes AM, Cheung SW
Generated figures and analyzed data: Aziz C
Wrote the R scripts used, performed statistical analyses, and contributed to the manuscript: Turinsky AL
Provided intellectual contribution to the analytical pipeline: Brudno M
Assisted with study design and manuscript preparation: Choufani S
Is the principal investigator and was involved in all aspects of the study: Weksberg R
Have read and approved the manuscript: All authors
Availability of data and materialsThe microarray data will be made available upon request.
Financial support and sponsorshipThis research was funded by the Canadian Institutes of Health Research (CIHR), the Province of Ontario Neurodevelopmental Disorders (POND) Network, in partnership with the Ontario Brain Institute (OBI), and the Canadian Centre for Computational Genomics (C3G). Goodman SJ is supported by The Lymphoma and Leukemia Society of Canada. Bioinformatic analyses were supported in part by the Canadian Centre for Computational Genomics (C3G), part of the Genome Technology Platform (GTP), funded by Genome Canada through Genome Quebec and Ontario Genomics (AL Turinsky, M Brudno), and Genome Canada through Ontario Genomics (AL Turinsky, M Brudno and R Weksberg). Chung BHY and Yeung KS are supported by the Health and Medical Research Fund and the Society of Relief for Disabled Children of Hong Kong (SRDC).
Conflicts of interestThe authors declared that there are no conflicts of interest.
Ethical approval and consent to participateInformed written consent to participate in the study was obtained from all research participants (or parents/legal guardians for those under 16) according to the protocol approved by the Research Ethics Board of the Hospital for Sick Children (REB# 1000038847).
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
3. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet 2013;14:204-20.
4. Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum Mol Genet 2005;14:R47-58.
5. Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am j hum genet 2012;90:260-72.
6. Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 2011;478:519-23.
7. Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, et al. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet 2017;100:725-36.
8. Fahrner JA, Bjornsson HT. Mendelian disorders of the epigenetic machinery: postnatal malleability and therapeutic prospects. Hum Mol Genet 2019;28:R254-64.
9. Cytrynbaum C, Choufani S, Weksberg R. Epigenetic signatures in overgrowth syndromes: Translational opportunities. Am J Med Genet A 2019;181:491-501.
10. Butcher DT, Cytrynbaum C, Turinsky AL, Siu MT, Inbar-Feigenberg M, et al. CHARGE and kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am J Hum Genet 2017;100:773-88.
11. Chater-Diehl E, Ejaz R, Cytrynbaum C, Siu MT, Turinsky A, et al. New insights into DNA methylation signatures: SMARCA2 variants in Nicolaides-Baraitser syndrome. BMC Med Genomics 2019;12:105.
13. Torres IO, Fujimori DG. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr opin struct biol 2015;35:68-75.
14. Fuks F. DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev 2005;15:490-5.
15. Choufani S, Cytrynbaum C, Chung BH, Turinsky AL, Grafodatskaya D, et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun 2015;6:10207.
16. Aref-Eshghi E, Bend EG, Hood RL, Schenkel LC, Carere DA, et al. BAFopathies’ DNA methylation epi-signatures demonstrate diagnostic utility and functional continuum of Coffin-Siris and Nicolaides-Baraitser syndromes. Nat Commun 2018;9:4885.
17. Aref-Eshghi E, Bend EG, Colaiacovo S, Caudle M, Chakrabarti R, et al. Diagnostic utility of genome-wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am J Hum Genet 2019;104:685-700.
18. Kleefstra T. Disruption of the gene euchromatin histone methyl transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J Med Genet 2005;42:299-306.
19. Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G, et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet 2009;46:598-606.
20. Pontén F, Jirström K, Uhlen M. The Human Protein Atlas - a tool for pathology. J Pathol 2008;216:387-93.
21. Yatsenko SA, Brundage EK, Roney EK, Cheung SW, Chinault AC, et al. Molecular mechanisms for subtelomeric rearrangements associated with the 9q34.3 microdeletion syndrome. Hum Mol Genet 2009;18:1924-36.
22. Kleefstra T, de Leeuw N. Kleefstra Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; .
23. Cormier-Daire V, Molinari F, Rio M, Raoul O, de Blois MC, et al. Cryptic terminal deletion of chromosome 9q34: a novel cause of syndromic obesity in childhood? J Med Genet 2003;40:300-3.
24. Stewart DR, Huang A, Faravelli F, Anderlid BM, Medne L, et al. Subtelomeric deletions of chromosome 9q: A novel microdeletion syndrome. Am J Med Genet A 2004;128A:340-51.
25. Yatsenko SA. Deletion 9q34.3 syndrome: genotype-phenotype correlations and an extended deletion in a patient with features of Opitz C trigonocephaly. J Med Genet 2005;42:328-35.
26. Willemsen MH, Vulto-van Silfhout AT, Nillesen WM, Wissink-Lindhout WM, van Bokhoven H, et al. Update on Kleefstra Syndrome. Mol Syndromol 2012;2:202-12.
27. Team RC. R: A language and environment for statistical computing. Vienna, Austria; 2013. Available from: https://www.R-project.org. [Last accessed on 25 May 2020].
28. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, et al. Discovery of cross-reactive probes and polymorphic CpGs in the illumina infinium HumanMethylation450 microarray. Epigenetics 2013;8:203-9.
29. Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014;30:1363-9.
30. Houseman EA, Houseman E, Accomando WP, Koestler DC, Christensen BC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC bioinformatics 2012;13:86.
31. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47.
33. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. Gene ontology: tool for the unification of biology. Nat Genet 2000;25:25-9.
34. Lee H, Graham JM, Rimoin DL, Lachman RS, Krejci P, et al. Exome sequencing identifies PDE4D mutations in acrodysostosis. Am J Hum Genet 2012;90:746-51.
35. Campbell CL, Collins RT II, Zarate YA. Severe neonatal presentation of Kleefstra syndrome in a patient with hypoplastic left heart syndrome and 9q34.3 microdeletion. Birth Defects Res A Clin Mol Teratol 2014;100:985-90.
36. Yatsenko SA, Hixson P, Roney EK, Scott DA, Schaaf CP, et al. Human subtelomeric copy number gains suggest a DNA replication mechanism for formation: beyond breakage-fusion-bridge for telomere stabilization. Hum Genet 2012;131:1895-910.
37. Schwaibold EMC, Smogavec M, Hobbiebrunken E, Winter L, Zoll B, et al. Intragenic duplication of EHMT1 gene results in Kleefstra syndrome. Mol Cytogenet 2014;7:74.
38. Ferry L, Fournier A, Tsusaka T, Adelmant G, Shimazu T, et al. Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol Cell 2017;67:550-65.e5.
39. Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007;317:1760-4.
40. von Meyenn F, Iurlaro M, Habibi E, Liu NQ, Salehzadeh-Yazdi A, et al. Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol Cell 2016;62:848-61.
41. Krzyzewska IM, Maas SM, Henneman P, Lip KVD, Venema A, et al. A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin epigenet 2019;11:156-15.
42. Aref-Eshghi E, Kerkhof J, Pedro VP, Barat-Houari M, Ruiz-Pallares N, et al. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 mendelian neurodevelopmental disorders. Am J Hum Genet 2020;106:356-70.
43. Hayashi S, Takeichi M. Emerging roles of protocadherins: from self-avoidance to enhancement of motility. J Cell Sci 2015;128:1455-64.
44. El Hajj N, Dittrich M, Haaf T. Epigenetic dysregulation of protocadherins in human disease. Semin Cell Dev Biol 2017;69:172-82.
45. Strong E, Butcher DT, Singhania R, Mervis CB, Morris CA, et al. Symmetrical dose-dependent DNA-methylation profiles in children with deletion or duplication of 7q11.23. Am J Hum Genet 2015;97:216-27.
46. Iacono G, Dubos A, Meziane H, Benevento M, Habibi E, et al. Increased H3K9 methylation and impaired expression of Protocadherins are associated with the cognitive dysfunctions of the Kleefstra syndrome. Nucleic Acids Res 2018;46:4950-65.
47. Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J 2008;27:2681-90.
48. Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, et al. Age-associated DNA methylation in pediatric populations. Gen Res 2012;22:623-32.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Goodman SJ, Cytrynbaum C, Chung BY, Chater-Diehl E, Aziz C, Turinsky AL, Kellam B, Keller M, Ko JM, Caluseriu O, Grafodatskaya D, McCready E, Perrier R, San Yeung K, Ho-Ming L, Machado J, Brudno M, Stavropoulos DJ, Scherer SW, Innes AM, Cheung SW, Choufani S, Weksberg R. EHMT1 pathogenic variants and 9q34.3 microdeletions share altered DNA methylation patterns in patients with Kleefstra syndrome. J Transl Genet Genom 2020;4:144-58. http://dx.doi.org/10.20517/jtgg.2020.23
AMA Style
Goodman SJ, Cytrynbaum C, Chung BY, Chater-Diehl E, Aziz C, Turinsky AL, Kellam B, Keller M, Ko JM, Caluseriu O, Grafodatskaya D, McCready E, Perrier R, San Yeung K, Ho-Ming L, Machado J, Brudno M, Stavropoulos DJ, Scherer SW, Innes AM, Cheung SW, Choufani S, Weksberg R. EHMT1 pathogenic variants and 9q34.3 microdeletions share altered DNA methylation patterns in patients with Kleefstra syndrome. Journal of Translational Genetics and Genomics. 2020; 4(3): 144-58. http://dx.doi.org/10.20517/jtgg.2020.23
Chicago/Turabian Style
Goodman, Sarah J., Cheryl Cytrynbaum, Brian Hon-Yin Chung, Eric Chater-Diehl, Celine Aziz, Andrei L. Turinsky, Barbara Kellam, Melanie Keller, Jung Min Ko, Oana Caluseriu, Daria Grafodatskaya, Elizabeth McCready, Renee Perrier, Kit San Yeung, Luk Ho-Ming, Jerry Machado, Michael Brudno, D. James Stavropoulos, Stephen W. Scherer, A. Micheil Innes, Sau Wei Cheung, Sanaa Choufani, Rosanna Weksberg. 2020. "EHMT1 pathogenic variants and 9q34.3 microdeletions share altered DNA methylation patterns in patients with Kleefstra syndrome" Journal of Translational Genetics and Genomics. 4, no.3: 144-58. http://dx.doi.org/10.20517/jtgg.2020.23
ACS Style
Goodman, SJ.; Cytrynbaum C.; Chung B.Y.; Chater-Diehl E.; Aziz C.; Turinsky AL.; Kellam B.; Keller M.; Ko JM.; Caluseriu O.; Grafodatskaya D.; McCready E.; Perrier R.; San Yeung K.; Ho-Ming L.; Machado J.; Brudno M.; Stavropoulos DJ.; Scherer SW.; Innes AM.; Cheung SW.; Choufani S.; Weksberg R. EHMT1 pathogenic variants and 9q34.3 microdeletions share altered DNA methylation patterns in patients with Kleefstra syndrome. J. Transl. Genet. Genom. 2020, 4, 144-58. http://dx.doi.org/10.20517/jtgg.2020.23
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 31 clicks
Cite This Article 31 clicks

 Like This Article 7
likes
Like This Article 7
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.