
Advancement in the diagnosis of mitochondrial diseases

Abstract
Mitochondrial diseases are multi-systemic, heterogeneous groups of diseases that are associated with various neuromuscular problems, cardiovascular disorders, metabolic syndrome, cancer, and obesity. Mitochondrial diseases are due to mutations in mitochondrial DNA or nuclear DNA that can affect the assembly of the mitochondrial components and mitochondrial function. Typically, mitochondrial diseases can be inherited through an autosomal dominant, autosomal recessive or X-linked pattern of inheritance. To date, there are more than 100 mitochondrial diseases identified. However, clinical phenotype heterogeneity is a huge problem for the diagnosis of mitochondrial diseases, as patients with the same mutations exhibit different clinical symptoms. Also, the heteroplasmy/homoplasmy conditions complicate the diagnosis process. Here, in this review, we discuss these challenges and problems in mitochondrial disease diagnosis, focusing on the mutational profile of both primary and secondary mitochondrial diseases. We also review the utilization of next-generation technology and multi-omics strategy to improve the diagnosis. The discussion addresses the current evidence of those applications and the challenges that need some improvement for better diagnosis yield.
Keywords
Introduction
Mitochondrial diseases are caused by mutations in mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) that encodes mitochondrial components. Mitochondrial diseases are complex diseases involving multiple organ systems, and the symptoms include deafness, blindness, dementia, movement disorders, cardiovascular diseases, and renal dysfunction[1]. Neurological and neuromuscular syndromes are the most common symptoms of mitochondrial diseases[1,2]. In addition, cardiovascular diseases, endocrine disorders, and cancers may also be associated with mtDNA mutations[3,4]. To date, there are about 350 causal genes for mitochondrial diseases[5]. Since mitochondria play a significant role in energy production via the oxidative phosphorylation (OXPHOS) system and Krebs cycle, disruptions in their genome and protein functions may affect various important cellular processes such as fatty acid metabolism, pyrimidine biosynthesis, calcium homeostasis, cell signaling, beta-oxidation and heme biosynthesis[6].
Several factors are involved in determining disease onset, clinical symptoms, phenotypic variability, and mitochondrial penetrance[7]. One such factor is replicative or mitotic segregation that leads to several conditions known as homoplasmy and heteroplasmy, threshold effect, clonal expansion, and the mtDNA bottleneck[7]. The ratio of wild type and mutant mtDNA is important in determining disease onset and clinical symptoms. The vast majority of mtDNA mutations are present in the heteroplasmy condition, and only some mtDNA mutations are homoplasmic and deleterious. Mitochondria are randomly segregated; thus, there is a possibility that the daughter cells can shift from wild type to mutant and vice versa. A random genetic drift results in clonal expansion, leading to acceleration of mtDNA mutation rate[8]. Rapid segregation of mammalian heteroplasmy with homoplasmy mtDNA between generations suggests that the mtDNA bottleneck occurs during development. Subsequently, this condition leads to differences in heteroplasmy levels in different mature oocytes of a woman[1].
mtDNA diseases have five unique characteristics, based on the mtDNA disease pedigree[1,7]. First, mtDNA mutations are inherited maternally. Second, mutations arise as a result of several factors, including lack of histone protection, deficiency in DNA repair mechanisms, and increased levels of reactive oxygen species (ROS) through OXPHOS activities in the mitochondria. Pathogenic mtDNA mutations comprise rearrangement mutations and point mutations in genes affecting mitochondrial protein translation and causing specific OXPHOS defects. Moreover, replicative segregation of mitochondria gives rise to homoplasmy and heteroplasmy conditions. These conditions cause differential phenotypes in various processes such as the transcription, translation, enzyme complex formation, respiratory complexes, biochemical levels, and cellular phenotypes[1,6,7,9]. Third, the impact of mtDNA mutations is subject to the amount of mitochondrial ATP production. In this case, tissues with the highest requirements for ATP can be affected the most, such as the central nervous system (CNS). Fourth, mtDNA repairs occur synonymously and replace mutations rapidly, about 5-10 times faster than nuclear OXPHOS genes due to mammalian mtDNA genes evolving faster compared to a single copy of nDNA. Finally, aging could result in decreased OXPHOS activities as well as mitochondrial dysfunction, which could be due to an accumulation of somatic cell mtDNA mutations[10].
The complexity and multi-systemic involvement in mitochondrial diseases render early diagnosis difficult. With ongoing advances in next-generation sequencing (NGS), the early diagnosis of mitochondrial diseases becomes feasible, and accurate diagnosis can be made even before the symptoms occur. In this review, we discuss the mitochondrial diseases, the challenges in their diagnosis, and future recommendations to assist in the diagnosis. We also provide the details of several companies that offer NGS services to diagnose mitochondrial diseases.
Overview of mtDNA and mitochondrial diseases
mtDNA
Mitochondria have their own genome, which is known as mtDNA[11]. The first complete sequence of the human mitochondrial genome was published in 1981[12]. Following that, in 1999, the mtDNA sequence was revised, and its final full genomic sequence was published[13]. In humans, mtDNA spans about 16,500 bp and consists of the heavy and light strands[12,13]. The heavy strand is rich in guanine bases and encodes 12 subunits of the oxidative phosphorylation (OXPHOS) system, two ribosomal RNAs (12S and 16S), and 14 tRNAs. The light strand encodes one subunit of OXPHOS and eight tRNAs. Altogether, mtDNA contains 37 genes encoding two rRNAs, 22 tRNAs, and 13 proteins subunits[12,13]. To date, there are about 1000-2000 mitochondrial proteins; however, only 600 of these proteins have determined functions. mtDNA is a multicopy genome, ranging from just ~100 copies in sperm to > 100,000 copies in mature oocytes[14]. The mtDNA genomes can exist either in homoplasmy (all genomes have an identical mtDNA genotype) or heteroplasmy (a combination of genomes with different mtDNA genotypes). These conditions can lead to various diseases or a normal phenotype, known as the phenotypic threshold effect. The inheritance of mtDNA is strictly maternal since mitochondrial endonuclease degrades the mtDNA within paternal mitochondria after fertilization[1,7,15].
There are five complexes in the OXPHOS system, namely complexes I (NADH ubiquinone oxidoreductase), complex II (succinate ubiquinone oxidoreductase), complex III (ubiquinone-cytochrome c reductase), complex IV (cytochrome c oxidase) and complex V (ATP synthase) which is important for energy production[6,12]. Embedded in this system are two electron carriers, ubiquinone coenzyme Q and cytochrome C, which serve as electron transporters important for energy production. Leakage of electrons from complexes I and III in the electron transport chain (ETC) can lead to the formation of ROS and oxidative stress conditions[9]. Oxidative stress occurs where there is an imbalance between the production of ROS, such as superoxide anion, H2O2, and the antioxidant defense mechanism to remove these radical molecules[9]. Uncontrolled oxidative stress may induce various dysfunctions within the mitochondria and cells, which could then lead to disease development [Figure 1].
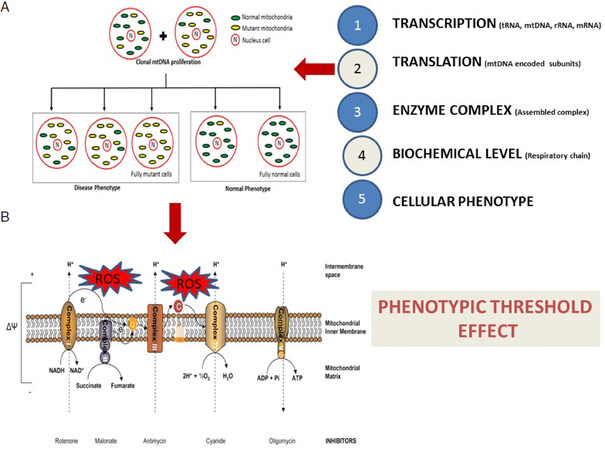
Figure 1. A: mtDNA mutations affect transcription, translation, enzyme complex, biochemistry, and cellular phenotype at different levels. These mutations may give rise to homoplasmy or heteroplasmy conditions, which can result in a disease or normal phenotype, known as a phenotypic threshold effect; B: The energy production process via oxidative phosphorylation (OXPHOS) may also lead to the formation of reactive oxygen species (ROS) in the electron transport chain. Oxidative stress conditions could further lead to the destruction of macromolecules, such as lipids, proteins, and DNA. Modified from a previous publication[16]
Different types of mitochondrial diseases
Mutations of mtDNA or nuclear DNA (nDNA) that encodes mitochondria components lead to an ineffective mitochondrial respiratory chain, and hence mitochondrial dysfunction[1,7,15]. The mitochondrial respiratory chain is an essential pathway for aerobic metabolism. Tissues and organs that are highly dependent on aerobic metabolism are prone to mitochondrial diseases[17]. Mitochondrial diseases can affect single or multiple organ systems[1,7,15], thus the association with various disease conditions including neurodegenerative conditions, cardiovascular diseases, neurometabolic problems, cancer and metabolic diseases such as obesity[3,4,10]. Mitochondrial diseases are considered rare, with a prevalence of 1:1,000,000. However, recent epidemiological studies suggested that at least 1 in 5000 individuals are affected by mitochondrial diseases[18,19]. Mitochondrial diseases may occur at any age[20], where mutations in mtDNA account for about 75% of adult diseases[18] but only about 25% of childhood-onset diseases[21,22]. Initially, it was postulated that nDNA abnormalities are present in childhood, while mtDNA abnormalities are only present in late childhood. Recently, many mtDNA diseases also present in childhood, and nDNA mutations are also present in adult life[23]. Mitochondrial diseases are classified into several categories, including primary and secondary mitochondrial diseases (SMD), which are discussed in detail in the next section [Figure 2].
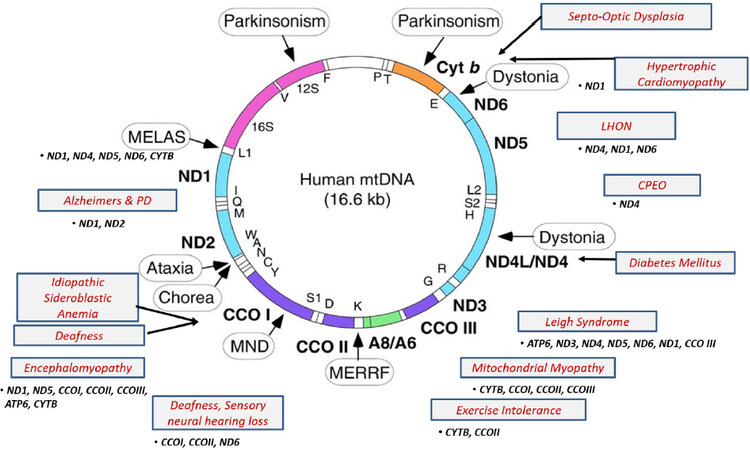
Figure 2. Common diseases associated with mtDNA mutations and the genes involved[24]. LHON: leber hereditary optic neuropathy; CPEO: chronic progressive external ophthalmoplegia; MERRF: myoclonic epilepsy with ragged-red fibers; MND: motor neuron disease; MELAS: mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; PD: Parkinson’s disease
Primary mitochondrial disease
As the mitochondrial components are inter-dependent of mtDNA and nDNA, most of the primary mitochondria diseases are due to these components. About 90% of mitochondrial proteins are from nuclear genes, which include the majority of the OXPHOS system. Also, mtDNA replication, transcription, and translation are dependent on these nuclear-encoded proteins. Primary mitochondrial diseases (PMD) are genetically inherited and diagnosed by identifying mutations in the mtDNA or nDNA[21], which could result in mitochondrial dysfunction [Tables 1 and 2]. Such examples are mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), neuropathy, ataxia and retinitis pigmentosa (NARP), Leigh syndrome, myoclonic epilepsy with ragged-red fibers (MERRF), Leber hereditary optic neuropathy (LHON), Kern-Sayre syndrome (KSS), mitochondrial neurogastrointestinal encephalopathy (MNGIE), and Alpers.
Summary of the most common mtDNA mutations in primary and secondary mitochondrial diseases
| Syndrome | Locus | Disease | Allele | Nt change | AA change | Ho | He | Ref. |
|---|---|---|---|---|---|---|---|---|
| Primary mitochondrial disease
Leigh syndrome/dystonia | ||||||||
| Leigh syndrome | MTATP6 | LS/NARP | T8993C | T-C | L156P | - | + | [48] |
| Leigh syndrome | MTATP6 | NARP | T8993G | T-G | L156R | - | + | [49,50] |
| Leigh syndrome | MTATP6 | LS | T9176G | T-G | L217R | + | + | [51] |
| Leigh syndrome | MTATP6 | LS/FBSN | T9176C | T-C | L217P | + | + | [52,53] |
| Leigh syndrome | MTATP6 | LS/ataxia/NARP-like disease | T9185C | T-C | L220P | + | + | [54] |
| Leigh syndrome | MTATP6 | LS | T9191C | T-C | L222P | - | + | [54] |
| Leigh syndrome | MTCO3 | LS-like | C9537insC | C-CC | Q111 frameshift | + | - | [55] |
| Leigh syndrome | MTND3 | LS | T10158C | T-C | S34P | + | + | [22,56,57] |
| Leigh syndrome | MTND3 | LS/LS-like disease/ESOC | T10191C | T-C | S45P | - | + | [58] |
| Leigh syndrome | MTND3 | LS/dystonia/stroke | G10197A | G-A | A-T | + | + | [59,60] |
| Leigh syndrome | MTND4 | LS | C11777A | C-A | R340S | - | + | [61,62] |
| Leigh syndrome | MTND5 | LS | T12706C | T-C | F124L | - | + | [63] |
| Dystonia | MTND1 | Adult-onset dystonia | A3796G | A-G | T164A | - | + | [64] |
| Dystonia/Leigh syndrome | MTND6 | LDYT/LS | G14459A | G-A | A72V | + | + | [65,66] |
| Dystonia/Leigh syndrome | MTND6 | LS/dystonia/Ataxia | T14487C | T-C | M63V | - | + | [67,68] |
| Leber hereditary optic neuropathy (LHON) | ||||||||
| Leber hereditary optic neuropathy | ND1 | LHON | G3460A | G-A | A52T | - | + | [69,70] |
| Leber hereditary optic neuropathy | ND4 | LHON | G11778A | G-A | R340H | - | + | [71] |
| Leber hereditary optic neuropathy | ND6 | LHON | T14484C | T-C | M64V | - | + | [69,72,73] |
| Encephalomyopathy | ||||||||
| Encephalomyopathy, MELAS | MTND1 | MELAS | T3308C | T-C | M1T | - | + | [74] |
| Encephalomyopathy, MELAS | MTND1 | MELAS/LHON | G3376A | G-A | E24K | - | + | [75] |
| Encephalomyopathy, MELAS | MTND1 | MELAS | G3697A | G-A | G131S | - | + | [60] |
| Encephalomyopathy, MELAS | MTND1 | MELAS | G3946A | G-A | E214K | + | + | [60] |
| Encephalomyopathy, MELAS | MTND1 | MELAS | T3949C | T-C | Y215H | - | + | [60] |
| Encephalomyopathy, MELAS | MTND4 | MELAS | A11084G | A-G | T109A | + | + | [76,77] |
| Encephalomyopathy, MELAS | MTND5 | MELAS | A12770G | A-G | E145G | - | + | [78] |
| Encephalomyopathy, MELAS | MTND5 | MELAS/LHON/LS overlap syndrome | A13045C | A-C | M237L | - | + | [78] |
| Encephalomyopathy, MELAS | MTND5 | MELAS/LS | A13084T | A-T | S250C | - | + | [79] |
| Encephalomyopathy, MELAS | MTND5 | MELAS/LS | G13513A | G-A | D393N | - | + | [80] |
| Encephalomyopathy, MELAS | MTND5 | MELAS | A13514G | A-G | D393G | - | + | [81] |
| Encephalomyopathy, MELAS | MTND6 | MELAS | G14453A | G-A | A74V | - | + | [82] |
| Encephalomyopathy, MELAS | MTCYB | MELAS/PD | 14787del4 | TTAA-del | I14frameshift | - | + | [83] |
| Encephalomyopathy, MELAS | MTTL1 | MELAS | A3243G | A-G | tRNALeu (UUR) | - | + | [84] |
| Encephalomyopathy, epilepsy | MTCO1 | Therapy-resistant epilepsy | C6489A | C-A | L196I | - | + | [85] |
| Encephalomyopathy, multisystem disorder | MTCO1 | Multisystem disorder | G6930A | G-A | G343Ter | - | + | [86] |
| Encephalomyopathy, multisystem disorder | MTCOI | Myopathy and cortical lesions | 6015del5 | Del 5 bp | Frameshift, 42 peptide | + | [87] | |
| Encephalomyopathy | MTCO2 | Encephalomyopathy | T7587C | T-C | M1T | - | + | [88] |
| Encephalomyopathy, multisystem disorder | MTCO2 | Multisystem disorder | G7896A | G-A | W104Ter | - | + | [89] |
| Encephalomyopathy, lactic acidosis | MTCO2 | Lactic acidosis | 8042del2 | AT-del | M153Ter | - | + | [90] |
| Encephalomyopathy | MTCO3 | Encephalomyopathy | G9952A | G-A | W248Ter | - | + | [91] |
| Encephalomyopathy, MELAS | MTCO3 | MELAS/PEM/NAION | T9957C | T-C | F251L | - | + | [92] |
| Encephalomyopathy, lactic acidosis | MTATP6 | Lactic acidosis/seizures | 9205del2 | TA-del | Ter227M | + | - | [93] |
| Encephalomyopathy, multisystem disorder | MTCYB | Multisystem disorder | A15579G | A-G | Y278C | - | + | [94] |
| Encephalomyopathy, septo-optic dysplasia | MTCYB | Septo-optic dysplasia | T14849C | T-C | S35P | - | + | [95] |
| Mitochondrial myopathy | ||||||||
| MM, exercise intolerance | MTCYB | EXIT | G14846A | G-A | G34S | - | + | [86] |
| MM | MTCYB | MM | G15059A | G-A | G190Ter | - | + | [96] |
| MM, exercise intolerance | MTCYB | EXIT | G15084A | G-A | W113Ter | - | + | [86] |
| MM, exercise intolerance | MTCYB | EXIT | G15150A | G-A | W135Ter | - | + | [97] |
| MM, exercise intolerance | MTCYB | EXIT | G15168A | G-A | W141Ter | - | + | [86] |
| MM, exercise intolerance | MTCYB | EXIT | T15197C | T-C | S151P | - | + | [97] |
| MM, exercise intolerance | MTCYB | EXIT/encephalomyopathy | G15242A | G-A | G166Ter | - | + | [98] |
| MM, exercise intolerance | MTCYB | EXIT | G15497A | G-A | G251S | + | - | [99] |
| MM, exercise intolerance | MTCYB | EXIT | 15498del24 | 24 bp deletion- | 251GDPDNYTL-del258 | - | + | [86] |
| MM, exercise intolerance | MTCYB | EXIT | G15615A | G-A | G290D | - | + | [100] |
| MM, exercise intolerance | MTCYB | EXIT | G15723A | G-A | W326Ter | - | + | [86] |
| Mitochondrial myopathy | MTCYB | MM | G15762A | G-A | G339E | - | + | [101] |
| MM, CPEO | MTND4 | CPEO | T11232C | T-C | L140P | - | + | [102] |
| MM, exercise intolerance | MTND4 | EXIT | G11832A | G-A | W358Ter | - | + | [86] |
| MM, exercise intolerance | MTCO1 | EXIT/myoglobinuria | G5920A | G-A | W6Ter | - | + | [103] |
| MM | MTCO1 | MM & rhabdomyolysis | G6708A | G-A | G269Ter | - | + | [104] |
| MM | MTCO2 | MM | T7671A | T-A | M29K | - | + | [105] |
| MM, exercise intolerance | MTCO2 | EXIT/rhabdomyolysis | T7989C | T-C | L135P | - | + | [106] |
| MM | MTCO3 | Myopathy and myoglobinuria | 9487del15 | Del 15 bp | Removed 5 aa | + | [107] | |
| MM | ND4 | MM | 3902inv7 bp | Inv 7 bp | D199G, L200K,A201V | + | - | [108] |
| Secondary mitochondrial diseases
Hypertrophic cardiomyopathy | ||||||||
| Hypertrophic cardiomyopathy | MTCYB | HCM | G15243A | G-A | G166E | - | + | [109] |
| Hypertrophic cardiomyopathy | MTCYB | HCM | G15498A | G-A | G251D | - | + | [110] |
| Diabetes mellitus | MTND4 | DM | A12026G | A-G | I423V | + | - | [74] |
| Idiopathic sideroblastic anemia | MTCO1 | SIDA | T6721C | T-C | M273T | - | + | [111] |
| Idiopathic sideroblastic anemia | MTCO1 | SIDA | T6742C | T-C | I280T | - | + | [111] |
| Deafness/sensorineural hearing loss | ||||||||
| Deafness | MTCO1 | DEAF | A7443G | A-G | Ter514G | + | - | [112] |
| Deafness | MTCO1 | DEAF | A7445C | A-C | Ter514S | + | - | [112] |
| Deafness, sensorineural hearing loss | MTCO1 | SNHL/LHON | G7444A | G-A | Ter514K | + | - | [112] |
| Deafness, sensorineural hearing loss | MTCO2 | SNHL | A8108G | A-G | I175V | + | - | [113] |
| Deafness, sensorineural hearing loss | MTND6 | SNHL | C14340T | C-T | V112M | + | - | [113] |
| Alzheimer’s and Parkinson’s diseases | ||||||||
| Alzheimer’s & Parkinson’s diseases | MTND1 | ADPD | A3397G | A-G | M31V | + | - | [114] |
| Alzheimer’s & Parkinson’s diseases | MTND2 | AD | G5460A | G-A | A331T | + | + | [115-117] |
| Alzheimer’s & Parkinson’s diseases | MTND2 | AD | G5460T | G-T | A331S | + | + | [115-117] |
Summary of known nuclear DNA mutations causing mitochondrial dysfunctions in primary and secondary mitochondrial diseases[118]
| Region | Gene/Locus | OMIM | Function | Chromosome | Inheritance | Clinical phenotype |
|---|---|---|---|---|---|---|
| Complex I | ACAD9 | 611103 | Assembly and activity | 3q26 | AR | Hypertrophic cardiopathy encephalopathy |
| FOXRED1 | 613622 | Assembly | 11q24.2 | AR | LS | |
| NUBPL | 613621 | Assembly | 14q12 | AR | Encephalomyopathy | |
| NDUFA1 | 300078 | HP fraction | Xq24 | X-linked | LS, progressive neurodegenerative disorder | |
| NDUFA2 | 602137 | HP fraction | 5q31.2 | AR | LS | |
| NDUFA9 | 603834 | HP fraction | 12p13.32 | AR | LS | |
| NDUFA10 | 603835 | HP fraction | 2q37.3 | AR | LS | |
| NDUFA11 | 612638 | IP fraction | 19p13.3 | AR | Fatal infantile lactic acidosis, encephalocardiomyopathy | |
| NDUFA12 | 609653 | HP fraction | 12q22 | AR | LS | |
| NDUFA13 | 609435 | HP fraction | 19p13.11 | AR | Encephalopathy, optic atrophy | |
| NDUFAF1(CIA30) | 606934 | Assembly | 15q13.3 | AR | Cardioencephalomyopathy | |
| NDUFAF2 (B17.2L) | 609653 | Assembly | 5q12.1 | AR | Early-onset progressive encephalopathy | |
| NDUFAF3 | 612911 | Assembly | 3p21.31 | AR | Neonatal encephalopathy | |
| NDUFAF4 (HRPAP2) | 611776 | Assembly | 6q16.1 | AR | Infantile encephalopathy | |
| NDUFAF5 (C20orf7) | 612360 | Assembly | 20p12.1 | AR | LS | |
| NDUFAF6 | 612392 | Assembly | 8q22.1 | AR | LS | |
| NDUFB3 | 603839 | HP fraction | 2q31.3 | AR | Fatal infantile lactic acidosis | |
| NDUFB9 | 601445 | HP fraction | 8q24.13 | AR | Hypotonia, lactic acidosis | |
| NDUFB10 | 603843 | HP fraction | 16p13.3 | AR | Lactic acidosis, cardiomyopathy | |
| NDUFB11 | 300403 | HP fraction | Xp11.3 | X-linked | Intrauterine growth restriction, lactic acidosis | |
| NDUFS1 | 157655 | IP fraction | 2q33-q34 | AR | LS | |
| NDUFS2 | 602985 | IP fraction | 1q23 | AR | Encephalopathy, cardiomyopathy | |
| NDUFS3 | 603846 | IP fraction | 11p11.11 | AR | LS | |
| NDUFS4 | 602694 | IP fraction | 5q11.1 | AR | LS | |
| NDUFS6 | 603848 | IP fraction | 5pter-p15.33 | AR | Fatal infantile lactic acidosis | |
| NDUFS7 | 601825 | HP fraction | 19p13.3 | AR | LS | |
| NDUFS8 | 602141 | HP fraction | 11q13 | AR | LS | |
| NDUFV1 | 161015 | FP fraction | 11q13 | AR | LS | |
| NDUFV2 | 600532 | FP fraction | 18p11 | AR | Cardiomyopathy, hypotonia, encephalopathy | |
| Complex II | SDH-A | 600857 | FP subunit | 5p15 | AR | LS |
| SDH-B | 185470 | IP subunit | 1p36.1-p35 | AD | Phaeochromocytoma and paraganglioma | |
| SDH-C | 602413 | Membrane subunit | 1q21 | AD | Autosomal dominant paraganglioma type 3 | |
| SDH-D | 602690 | Membrane subunit | 11q23 | AD | Autosomal dominant paraganglioma type 1, pheochromocytoma | |
| SDHAF1 | 612848 | Assembly | 19q12-q13.2 | AR | Leukoencephalopathy | |
| SDHAF2 | 613019 | Assembly | 11q12.2 | AD | Autosomal dominant paraganglioma type 2 | |
| Complex III | BCS1L | 603647 | Assembly | 2q33 | AR | Encephalopathy, hepatic failure and tubulopathy, LS, GRACILE syndrome, Bjornstad syndrome |
| UQCC2 | 614461 | Assembly | 6p21.31 | AR | Lactic acidosis and renal tubular dysfunction | |
| UQCC3 | 616097 | Assembly | 11q12.3 | AR | Lactic acidosis, hypoglycemia, hypotonia | |
| UQCRB | 191330 | Electron transfer | 8q22 | AR | Hypoglycemia, lactic acidosis | |
| UQCRQ | 612080 | Electron transfer | 5q31.1 | AR | Severe neurological phenotype | |
| Complex IV | COA3 | 614775 | Assembly | 17q21.2 | AR | Neuropathy, exercise intolerance |
| COA5 | 613920 | Assembly | 2q11.2 | AR | Cardioencephalomyopathy | |
| COA6 | 614772 | Assembly | 1q42.2 | AR | Cardioencephalomyopathy | |
| COX10 | 602125 | Heme A farnesyltransferase | 17p12-p11.2 | AR | Neonatal tubulopathy and encephalopathy, LS, cardiomyopathy | |
| COX14 (C12orf62) | 614478 | COX assembly | 12q13.12 | AR | Neonatal lactic acidosis | |
| COX15 | 603646 | Heme A synthesis | 10q24 | AR | Early-onset hypertrophic cardiomyopathy, LS | |
| COX20 | 614698 | Assembly | 1q44 | AR | Ataxia, muscle hypotonia | |
| COX6A1 | 602072 | Cytochrome oxidase activity | 12q24.31 | AR | Charcot-Marie-Tooth disease | |
| COX6B1 | 124089 | Cytochrome oxidase activity and assembly | 19q13.1 | AR | Encephalomyopathy | |
| COX7B | 300885 | Cytochrome oxidase activity | Xq21.1 | X-linked | Microphthalmia with linear skin lesions | |
| COX8A | 123870 | Cytochrome oxidase activity | 11q13.1 | AR | LS | |
| FASTKD2 | 612322 | Role in apoptosis | 2q33.3 | AR | Encephalomyopathy | |
| LRPPRC | 220111 607544 | Assembly | 2p21-p16 | AR | French-Canadian LS | |
| SCO1 | 603644 | Copper transport | 17p13-p12 | AR | Neonatal hepatic failure and encephalopathy | |
| SCO2 | 604272 | Copper transport | 22q13 | AR | Neonatal cardioencephalomyopathy | |
| SURF1 | 185620 | Assembly | 9q34 | AR | LS | |
| TACO1 | 612958 | Translational activator of COX1 | 17q22-q24.2 | AR | LS | |
| Complex V | ATP5E | 606153 | ATPase activity | 20q13.3 | AR | Lactic acidosis, mental retardation, peripheral neuropathy |
| ATP5A1 | 164360 | ATPase activity | 18q21.1 | AR | Neonatal encephalopathy | |
| ATP8A2 | 605870 | ATPase activity | 13q12.13 | AR | Cerebellar ataxia, mental retardation | |
| ATPAF2 | 608918 | Assembly | 17p11.2 | AR | Early-onset encephalopathy, lactic acidosis | |
| TMEM70 | 604273 612418 | Assembly | 8q21.11 | AR | Neonatal encephalopathy, cardiomyopathy | |
| MtDNA maintenance | ANT1 (PEOA2) | 609283 | Adenine nucleotide translocator isoform 1 | 4q35 | AD-AR | AD-PEO, multiple mtDNA deletions |
| C10ORF (PEOA3) | 609286 | Twinkle helicase | 10q24 | AD | AD-PEO, SANDO syndrome | |
| DGUOK | 601465 | Deoxyguanosine kinase Mitochondrial dNTP pool maintenance | 2p13 | AR | Hepatocerebral mtDNA depletion syndrome | |
| FBXL4 | 605654 | mtDNA maintenance | 6q16.1-q16.2 | AR | Encephalomyopathy and myopathy, mtDNA depletion | |
| MFN2 | 609260 | Mitofusin, mitochondrial fusion | 1p36-p35 | AD | Charcot-Marie-Tooth disease-2A2 (CMT2A2), multiple deletions | |
| MGME1 | 615084 | mtDNA maintenance | 20p11.23 | AR | CPEO and myopathy, mtDNA depletion | |
| MPV17 | 137960 | Regulation of mtDNA copy number | 2p23-p21 | AR | Hepatocerebral MDDS | |
| OPA1 | 165500 | Dynamin-related protein | 3q28-q29 | AD | AD-optic atrophy, multiple deletions | |
| POLG (PEOA1) | 174763 | Polymerase gamma mtDNA replication | 15q25 | AD-AR | Alpers syndrome, AD-PEO, and AR-PEO, male infertility, SANDO* syndrome, SCAE* | |
| POLG2 (PEOA4) | 610131 | Catalytic subunit of DNA polymerase gamma | 17q23-q24 | AD | AD-PEO | |
| RRM2B (PEOA5) | 604712 | Ribonucleotide reductase M2 B dNTP pool | 8q23.1 | AR | Encephalomyopathy, renal tubulopathy MNGIE, AD-PEO | |
| SUCLA2 | 603921 | Succinate-CoA ligase, ADP-forming, beta subunit | 13q12.2-q13 | AR | Encephalomyopathy with methylmalonic aciduria | |
| SUCLG1 | 611224 | Succinate-CoA ligase, alpha subunit | 2p11.2 | AR | Encephalomyopathy with methylmalonic aciduria | |
| TFAM | 600438 | Mitochondrial transcription factor A | 10q21.1 | AR | Encephalomyopathy, mtDNA depletion | |
| TK2 | 188250 | Thymidine kinase Mitochondrial dNTP pool maintenance | 16q22 | AR | Myopathic mtDNA depletion | |
| TYMP (ECGF1) | 603041 | Thymidine phosphorylase | 22q13.32-qter | AR | MNGIE, mtDNA depletion | |
| Mitochondrial import | DNAJC19 | 608977 | Protein import | 3q26.3 | AR | Cardiomyopathy, ataxia |
| DDP | 304700 | Protein import | Xq22 | X-linked | Deafness-dystonia or Mohr-Tranebjaerg syndrome | |
| Mitochondrial protein synthesis | AARS2 | 612035 | Alanyl-tRNA synthetase | 6p21.1 | AR | Cardiomyopathy, leukoencephalopathy |
| CARS2 | 612800 | Cysteinyl-tRNA synthetase | 13q34 | AR | Myoclonic epilepsy | |
| C12orf65 | 613541 | Mitochondrial translation | 12q24.31 | AR | Encephalomyopathy, optic atrophy, axonal neuropathy, paraparesis | |
| DARS2 | 611105 | Aspartyl-tRNA synthetase | 1q25.1 | AR | Leukoencephalopathy and lactic acidosis | |
| EARS2 | 612799 | Glutamyl tRNA synthetase | 16p12.2 | AR | Leukoencephalopathy | |
| EFG1 | 609060 | Elongation factor G1 mitochondrial translation defect | 3q25 | AR | Severe hepato-encephalopathy and lactic acidosis | |
| FARS2 | 611592 | Phenylalanyl-tRNA synthetase | 6p25.1 | AR | Alpers syndrome, spastic paraplegia | |
| GARS | 600287 | Glycyl-tRNA synthetase | 7p14.3 | AD | Charcot-Marie-Tooth disease | |
| GFM1 | 606639 | Mitochondrial translation elongation | 3q25.32 | AR | Encephalopathy/hepatic failure | |
| GFM2 | 606544 | Mitochondrial translation elongation | 5q13.3 | AR | Neurodevelopmental disorder, dysmorphic features | |
| GTPBP3 | 608536 | GTP-binding protein | 19p13.11 | AR | Cardiomyopathy, encephalopathy | |
| HARS2 | 600783 | Histidyl-tRNA synthetase | 5q31.3 | AR | Perrault syndrome | |
| IARS2 | 612801 | Isoleucyl tRNA-Synthetase | 1q41 | AR | Cataract, deafness, neuropathy/Leigh syndrome | |
| KARS | 601421 | Lysyl-tRNA synthetase | 16q23.1 | AR | CMT disease/deafness | |
| LARS | 615438 | Leucine-tRNA synthetase | 5q32 | AR | Hepatopathy | |
| LARS2 | 604544 | Leucyl-tRNA Synthetase | 3p21.31 | AR | Perrault syndrome | |
| MRPL3 | 607118 | Mitochondrial translation | 3q22.1 | AR | Cardiomyopathy, mental retardation | |
| MRPS7 | 611974 | Mitochondrial translation | 17q25.1 | AR | Deafness, hepatic and renal failure | |
| MRPL12 | 602375 | Mitochondrial translation | 17q25.3 | AR | Growth retardation, encephalopathy | |
| MRPS16 | 609204 | Mitochondrial translation | 10q22.1 | AR | Neonatal lactic acidosis, corpus callosum agenesis | |
| MRPS22 | 605810 | Mitochondrial translation | 3q23 | AR | Cardiomyopathy, tubulopathy | |
| MRPL44 | 611849 | Mitochondrial translation | 2q36.1 | AR | Cardiomyopathy | |
| MTFMT | 611766 | Mitochondrial translation | 15q22.31 | AR | LS | |
| MTO1 | 614667 | tRNA modification | 6q13 | AR | Cardiomyopathy | |
| NARS2 | 612803 | Asparaginyl-tRNA synthetase | 11q14.1 | AR | Alpers syndrome/nonsyndromic deafness and Leigh syndrome | |
| PARS2 | 612036 | Prolyl- tRNA Synthetase | 1p32.3 | AR | Alpers syndrome | |
| RARS2 | 611523 | Arginyl-tRNA synthetase | 6q16.1 | AR | Pontocerebellar hypoplasia | |
| RMND1 | 614917 | Mitochondrial translation | 6q25.1 | AR | Encephalopathy | |
| SARS2 | 612804 | seryl-tRNA synthetase | 19q13.2 | AR | Hyperuricemia, pulmonary hypertension, renal failure | |
| TARS2 | 612805 | Threonyl--tRNA synthetase | 1q21.2 | AR | Encephalomyopathy | |
| TSFM | 604723 | Mitochondrial translation elongation | 12q13-q14 | AR | Encephalomyopathy, hypertrophic cardiomyopathy | |
| TUFM | 602389 | Mitochondrial translation elongation | 16p11.2 | AR | Leukodystrophy with micropolygyria | |
| TRMT5 | 611023 | Mitochondrial tRNA methylation | 14q23.1 | AR | Cardiomyopathy/exercise intolerance | |
| TRMT10C | 615423 | tRNA methyltransferase | 3q12.3 | AR | Hypotonia, feeding difficulties, deafness | |
| TRMU | 610230 | Mitochondrial translation | 22q13.31 | AR | Liver failure, deafness | |
| VARS2 | 612802 | Valyl-tRNA synthetase | 6p21.33 | AR | Encephalomyopathy | |
| YARS2 | 610957 | yrosyl-tRNA synthetase | 12p11.21 | AR | Myopathy, lactic acidosis, and sideroblastic anemia-2 | |
| Iron homeostasis | ABCB7 | 301310 | Iron transport | Xq13.1-q13.3 | X-linked | X-linked sideroblastic anemia with ataxia |
| BOLA3 | 613183 | Iron-sulfur cluster biosynthesis | 2p13.1 | AR | Encephalomyopathy, cardiomyopathy | |
| FDXL1 | 614585 | Iron-sulfur cluster biosynthesis | 19p13.2 | AR | Myopathy, lactic acidosis | |
| FRDA (FXN) | 606829 | Frataxin trinuc.* repeat, | 9q13 | AR | Friedreich ataxia, neuropathy, cardiomyopathy, diabetes | |
| GLRX5 | 205950 | Iron-sulfur cluster biosynthesis | 3p22.1 | AR | Sideroblastic anemia | |
| IBA57 | 615316 | Iron-sulfur cluster biosynthesis | 1q42.13 | AR | Myopathy, encephalopathy | |
| ISCA2 | 615317 | Iron-sulfur cluster biosynthesis | 14q24.3 | AR | Leukodystrophy | |
| ISCU | 255125 | Iron-sulfur cluster biosynthesis | 12q23.3 | AR | Myopathy, lactic acidosis, exercise intolerance | |
| LYRM4 | 613311 | Iron-sulfur cluster biosynthesis | 6p25.1 | AR | Lactic acidosis, Failure to thrive | |
| LYRM7 | 615831 | Iron-sulfur cluster biosynthesis | 5q23.3-q31.1 | AR | Encephalopathy, lactic acidosis | |
| NFU1 | 608100 | Iron-sulfur cluster biosynthesis | 2p13.3 | AR | Lactic acidosis, multiple respiratory chain deficiency | |
| Coenzyme Q10 biogenesis | APTX | 606350 | CoQ10 deficiency | 9p13.3 | AR | Cerebellar ataxia, Oculomotor apraxia |
| CABC1 | 606980 | CoQ10 deficiency | 1q42.2 | AR | Cerebellar ataxia, lactic acidosis | |
| COQ2 | 609825 | CoQ10 deficiency | 4q21-q22 | AR | Encephalomyopathy, nephropathy | |
| COQ4 | 612898 | CoQ10 deficiency | 9q34.13 | AR | Encephalomyopathy, mental retardation | |
| COQ5 | 616359 | CoQ10 deficiency | 12q24.31 | AR | Encephalomyopathy, cerebellar ataxia | |
| COQ6 | 614647 | CoQ10 deficiency | 14q24.3 | AR | Nephrotic syndrome, deafness | |
| COQ7 | 601683 | CoQ10 deficiency | 16p12.3 | AR | Hypotonia, cardiac hypertrophy | |
| COQ9 | 612837 | CoQ10 deficiency | 16q13 | AR | Neonatal lactic acidosis, seizures, cardiomyopathy | |
| PDSS1 | 607429 | CoQ10 deficiency | 10p12.1 | AR | Deafness, valvulopathy, mental retardation | |
| PDSS2 | 610564 | CoQ10 deficiency | 6q21 | AR | LS, nephrotic syndrome | |
| Chaperone function | HSPD1 | 118190 | Mitochondrial chaperone | 2q33.1 | AR | Spastic paraplegia, leukodystrophy |
| SPG7 | 607259 | Paraplegin ATPase protease | 16q24.3 | AR | Spastic paraplegia | |
| Mitochondrial integrity | DLP1 | 603850 | Mitochondrial and peroxisomal fission | 12p11.21 | AD | Microcephaly, abnormal brain development, optic atrophy, lactic acidosis |
| G4.5 (Tafazzin) | 302060 | Cardiolipin defect | Xq28 | X-linked | Barth syndrome, X-linked dilated cardiomyopathy | |
| RMRP | 250250 | RNAse Mitochondrial RNA processing | 9p13-p12 | AR | Metaphyseal chondrodysplasia or cartilage-hair hypoplasia | |
| Mitochondrial metabolism | ATAD3 | 617183 | Mitochondrial dynamics | 1p36.33 | AR/AD | Neurodevelopmental disorder, pontocerebellar hypoplasia, encephalopathy |
| ETHE1 | 602473 | Ethylmalonic acid metabolism | 19q13 | AR | Encephalopathy, ethylmalonic aciduria | |
| PDHA1 | 308930 | Pyruvate dehydrogenase E1-a subunit | Xp22.2-p22.1 | X-linked | LS | |
| PUS1 | 600462 | Pseudouridine synthase | 12q24.33 | AR | Myopathy, lactic acidosis, and sideroblastic anemia |
MELAS disease is diagnosed by the presence of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. The m.3243A>G mutation in the MT-TL1 gene encoding tRNALEU(UUR) was identified to be the cause of MELAS in 1990. This mutation causes translational defects of the OXPHOS protein assembly and the lack of complex I (CI), which leads to an accumulation of nicotinamide adenine dinucleotide (NAD)+ hydrogen (H) (NADH), increased glycolysis, oxidative stress, and reduced ATP production[25]. Similarly, NARP disease is also associated with energy production due to a mutation in the ATP6 gene (m.9176 T>G)[26]. This mutation leads to a heterogeneous and complex disease manifestation, from having adult-onset NARP to fatal infantile subacute necrotizing encephalomyopathy[27]. A study performed by Mordel and colleagues showed that a novel and pathogenic 2-bp microdeletion (m9127-9128 del AT) in the ATP6 gene caused NARP[28]. This heteroplasmy mutation was highly abundant in the muscle (82%), followed by fibroblasts (50%) and blood (10-20%). Functional studies showed that oligomycin-sensitive ATPase hydrolytic activity was reduced at 60%, and ATP synthesis was decreased at 40%[28].
The most common mitochondrial disease in children is Leigh syndrome, with a prevalence of 1:40,000[29]. The diagnosis is based on neuroimaging and characterized by symmetrical lesions in the basal ganglia, thalamus, and brain stem. Patients with Leigh syndrome usually present with loss of acquired cognitive, visual as well as motor skills. Mutations in both mtDNA and nDNA could lead to mitochondrial dysfunctions in different aspects. For example, mutations in NDUFS2 and SURF1 (structural subunits and assembly factors), PHHA1 (Kreb’s cycle components), MTFMT (mitochondrial protein translation), and ECHSI (valine metabolism), are all reported to be associated with Leigh syndrome[30]. Another similar disease is MERRF, which affects 1 in 400,000 individuals and involves the nervous and skeletal muscle systems. MERRF is characterized by myoclonus epilepsy, ataxia, seizures, and myopathy symptoms. The most common mutation is the mtDNA tRNALys A8344G, which accounts for about 80%-90% of all MERRF cases[31]. Another common mutation, causing LHON disease, occurs 1 in 31,000-50,000 people. Clinically, it is characterized by bilateral loss of central vision and some other abnormalities such as movement disorders, dystonia, or multiple sclerosis-like symptoms. Mutations in the NADH dehydrogenase account for 90% of all LHON cases, and the most common mutations are m.11778G>A (ND4), m.14484T>C (ND6) and m.3460G>A (ND1)[32,33]. LHON disease can lead to blindness due to the loss of retinal ganglion cells from the mtDNA mutations affecting the respiratory complex I (CI) subunits, such as m.3460G>A, m.11778G > A and m.14484 T>C. There is also a rare mutation such as m.13094T>C reported in LHON disease[34].
Another PMD with eye disorder is the Kern-Sayre syndrome (KSS), which is characterized by a progressive external ophthalmoplegia, a condition that causes eye muscle paralysis. The patients may also have other symptoms such as cardiac conduction defect, ataxia, and abnormalities in protein levels of the cerebrospinal fluid. The disease affects 1-3 in 100,000 individuals, and most KSS cases are due to mtDNA mutations. Some of the mutations that have been identified in KSS are the deletion of 3,236 bp of mitochondrial chromosome from 10,170[35], mtDNA duplication[36], a mtDNA point mutation in the MTTL2 gene[37], and a large mtDNA deletion[38]. Mitochondrial neurogastrointestinal encephalopathy (MNGIE) disease affects several parts of the body, mostly the digestive and nervous systems[39]. It is caused by a mutation in the thymidine phosphorylase gene (TYMP) that leads to decreased enzyme activity and increased thymidine levels[40]. MNGIE is a progressive multi-system disorder with various symptoms, including dysphagia, gastroesophageal reflux disease, gastroparesis, intestinal pseudo-obstruction, muscle weakness, ptosis, and ophthalmoparesis. This condition affects intra-mitochondrial nucleoside levels leading to mtDNA depletion and secondary mtDNA mutations[41]. Mutations in the POLG and RRM2B genes have also been described in MNGIE-type phenotypes[42,43].
Another PMD is Alpers disease, which is a childhood progressive neurological disorder that affects 1 in 100,000 individuals. The clinical characteristics of Alpers disease include recurrent seizures, mental loss, movement disabilities, and liver disease with clinical symptoms of spasticity, seizures, and dementia. POLG gene mutations occur in 13% of Alpers cases, resulting in reduced DNA replication, mtDNA depletion, and subsequently reduced ATP production[44]. mtDNA depletion syndrome (MDDS) is an autosomal recessive disease that is characterized by severe depletion of mtDNA in tissues. MDDS disease is clinically heterogeneous, in which affected tissues can be in a single organ or multiple organs, including liver, brain, kidney, and muscles. The known cause of MDDS is due to mutations in nuclear genes (nDNA) that affect mtDNA maintenance [Table 2]. The known genes are those that encode thymidine kinase 2 (TK2), guanosine diphosphate (GDP)-forming succinyl CoA ligase alpha subunit (SUCLG1), adenosine diphosphate (ADP)-forming succinyl CoA ligase beta subunit (SUCLA2), RRM2B, TYMP and deoxyguanosine kinase (DGUOK). All of these genes are responsible for maintaining the mitochondrial dNTP pools during mtDNA synthesis[45]. In some cases, mutations in POLG and Twinkle (TWNK) that are involved in mtDNA replication will also result in insufficient mtDNA synthesis[45,46]. Similar to MDDS, ataxia neuropathy spectrum (ANS) is also due to mutations in nuclear genes (nDNA), namely POLG and TWNK genes. Patients with ANS typically have symptoms involving nerve, brain, and muscle dysfunctions. Currently, patients with mitochondrial recessive ataxia syndrome (MIRAS) and sensory ataxia neuropathy dysarthria and ophthalmoplegia (SANDO) are considered having ANS. Mutations in POLG are often inherited via an autosomal recessive pattern, whereas mutations in TWNK are often inherited in an autosomal dominant pattern[47].
Secondary mitochondrial diseases
Secondary mitochondrial diseases (SMD) constitute a group of complex disease manifestations that can be caused by genetic as well as environmental factors. From a genetic point of view, SMD could be due to pathogenic mutations in other genes that are not related to OXPHOS components, but these mutations disrupt mitochondrial function. Also, adverse environmental effects or other factors such as oxidative stress, aging, drug mitotoxicity, and inflammation are some of the processes that could alter mitochondrial functions. If PMD is inherited, then SMD could be inherited and acquired. Typically, SMD occur after conception, which could result in dysfunction in mitochondrial ATP and also non-ATP (mitochondrial fission and fusion) producing capabilities. Mitochondria are highly dynamic organelles and undergo mitochondrial fission and fusion processes regularly[119]. Fission is a process of making short, rod- or sphere-shaped mitochondria controlled by DRP1, which is a large GTPase of the dynamin superfamily[119]. In contrast, fusion leads to the formation of long and filamentous mitochondria, involving MFN1, MFN2, and OPA1[119]. In normal conditions, mitochondrial dynamics are well-maintained; however, oxidative stress (intra- and extracellular stresses) could disrupt this balance, generating fragmented mitochondria.
Mitochondrial dynamics maintenance is important to preserve mitochondrial shapes, as well as its functions, inheritance, quality control, and cellular apoptosis[119]. Inefficient mitochondrial dynamics could lead to multifactorial diseases, including diabetes, cancer, and kidney and neurodegenerative diseases[120]. Distinguishing whether mitochondrial dysfunction is inherited or acquired is extremely challenging and still poorly understood[121]. Also, PMD and SMD can have overlapping phenotypes or symptoms; moreover, some patients may not have all the components of mitochondrial disease criteria (MDC). Currently, MDC is used to differentiate between PMD and SMD[122]. MDC uses several criteria, including energy production, biochemical, clinical, tissue, and molecular characterizations. However, there are no universal guidelines to diagnose mitochondrial diseases worldwide. Advances in NGS may assist in the diagnosis of PMD or SMD accurately[123]. Comprehensive molecular profiling could determine which genes and pathways are related to PMD and SMD, thereby improving the diagnosis of PMD or SMD.
Mitochondria and diabetes
Energy production through OXPHOS process in the mitochondria may also lead to oxidative stress conditions by increasing ROS formation in the cells. ROS may activate pro-inflammatory pathways, reduce nitric oxide bioavailability, and could further induce diabetic endothelial dysfunction, subsequently leading to the development of diabetes and diabetic kidney disease (DKD)[124]. Diabetes is an endocrine disorder due to multiple factors, including genetics, impaired insulin action, obesity, inflammation, impaired autophagy, increased oxidative stress, and mitochondrial dysfunction[124]. The relationship of diabetes with oxidative stress and mitochondrial dysfunction can partially be explained by the damage-associated molecular pattern (DAMPs) that could initiate inflammatory response via various pathways such as T signaling pathways by interacting with (1) Toll-like receptors (TLRs), (2) nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, and (3) cytosolic cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) DNA-sensing system[120]. Interestingly, Li and colleagues have identified that m.15897G>A mutation of tRNAThr, which belongs to the haplogroup D4b1, is present in Type 2 diabetes Chinese patients, and this mutation was maternally inherited[125]. The functional study of this mutation showed that a decreased efficiency of mitochondrial tRNAThr leads to reduced efficiency of OXPHOS protein synthesis and assembly and ATP synthesis, and decreased mitochondrial membrane potential (MMP)[125].
Insulin resistance (IR) is one of the main risk factors for type 2 diabetes, and mitochondrial dysfunction is related to IR development. Recent in vivo and ex vivo metabolic studies involving humans and rodents showed that mitochondrial dysfunction could lead to ectopic lipid deposition and IR[126]. Pereira and colleagues showed that mtDNA could activate the NLRP3 inflammasome, which subsequently causes endothelial dysfunction and inflammation in diabetes. Diabetes reduces endothelium-dependent vasodilation and escalates vascular ROS generation and caspase-1 and IL-1β activation in streptozotocin (STZ)-induced diabetic C57BL/6 mice, but not in those Nlrp3-/-. Deficiency in NLRP3 could prevent diabetes-associated vascular inflammatory damage and endothelial dysfunction[127]. Another example of how mitochondrial dysfunction is associated with diabetes is via the action of the anti-diabetic drug metformin (MF). The protective effect of MF on regulatory networks and integrated stress responses was observed in the brain tissue of STZ-induced diabetic mice. STZ-induced diabetic mice treated with MF (20 mg/kg) showed a significant decrease in protein carbonylation and oxidation. MF treatment also improved mitochondrial function via the increase of the chaperone proteins (HSP60, HSP70, and LonP1)[128]. However, the exact mechanisms of how mtDNA causes diabetes are still not fully understood.
Mitochondria and cardiovascular diseases
The role of mtDNA mutations in cardiovascular diseases (CVD) has been discussed extensively[3]. The MtDNA control region is important for controlling mtDNA gene expression. Umbria and colleagues studied mutations in the mtDNA control region in 154 stroke cases and 211 myocardial infarction (MI) patients[129]. They found that the m.16145 G>A and m.16311 T> C variants could be risk factors for stroke (conditional logistic regression, P = 0.038 & P = 0.018, respectively), and that the m.72 T>C and m.73 A>G variants may be protective against MI (conditional logistic regression, P = 0.001 & P = 0.009, respectively)[129]. ROS molecules are very harmful and may damage macromolecules, such as proteins, lipids and DNA[130]. One such mechanism for ROS effects is the accumulation of damage-associated molecular patterns (DAMPs), which could activate pathogen recognition receptors (PRRs), triggering chronic inflammation-induced oxidative damage[120]. Several pathways associated with DAMPS’ action are the mitochondrial calcium handling ROS production, TLRs, NLRP3 inflammasome, cytosolic cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) DNA-sensing system, and nuclear factor kappa B (NF-kB)[120].
Another mechanism of how mitochondria could be associated with CVD is via circadian clock regulation[126]. Mitochondria are among the organelles that are important in controlling the crosstalk between the circadian clock and metabolic pathways, the intestinal microbiota, and the immune system as well. Mitochondria regulate circadian rhythmicity through NAD+ production, SIRT1/SIRT3 activation, and mitochondrial dynamics[131]. Yang and colleagues suggested that interference of the Clock gene could suppress mitochondrial apoptosis pathways by stabilizing mitochondrial membrane potential (MMP) and inhibiting mitochondrial membrane permeabilization. These could be due to reduced BAD and BIM proteins that are essential for apoptosis, as well as lower expression of mitochondrial apoptosis factors, i.e., AIF, CYCS, APAF-1, and SMAC, which suppress the formation of the apoptosome and DNA degradation[132]. The circadian clock plays a vital role in transcriptional-translational processes in cellular metabolism and mitochondrial activity[133]. Perturbations to circadian rhythm could lead to CVD[134-137], for example, the circadian clock genes such as BMAL1 can affect vascular proliferation[138], and CLOCK and ARNTL may be related to MI[139]. Zhang and colleagues also showed that the circadian rhythm and clock genes are related to acute coronary syndrome (ACS), in which plaque stability was negatively correlated with the expression levels of clock genes. The levels of MMP2 and MMP9 were increased in ST-segment elevation myocardial infarction, non-ST segment elevation myocardial infarction and unstable angina pectoris (UA) compared to the control group (P < 0.05)[140]. Also, Wang and colleagues showed that increased mtDNA 8-OHdG could increase the odds of having coronary artery disease (CAD) (OR = 1.38), coronary stenosis (OR = 1.29), and higher levels of C-reactive protein[141]. Although these findings supported the notion that mitochondrial dysfunction could contribute to the development of CVD, the exact mechanism for that association is still unknown.
Mitochondria and cancer
One of the hallmarks of cancer cell development is the metabolic changes known as the “Warburg effect” that shows the role of mitochondria in cancer[142,143]. The Warburg effect refers to the situation of cancer cells switching their metabolism and energy production from the oxidative phosphorylation (OXPHOS) to glycolysis with lactic acid production, despite the presence of oxygen (aerobic glycolysis)[144]. Although the glycolysis process produces less energy than OXPHOS, the abundance of the glucose influx in the cells can result in more energy production at a faster rate potentially. Initially, the Warburg effect is thought of as a result of the mitochondrial defects that inhibit OXPHOS, eventually causing cancer development[144]. However, recent findings showed that many cancer cells have functional mitochondria, in which some exhibit a high level of OXPHOS activity. Whereas some are more glycolytic but still retain their mitochondrial functions[4]. Moreover, the discovery of the oncogenes in cancer explains that this switching of metabolism or metabolic re-programming is a complex process and may be due to the activation of oncogenic genes.
Activation of the oncogenic driver mutations in KRAS, PI3K, AKT, mTOR, and MYC, as well as the loss of tumor suppressor expression such as p53, facilitate metabolic switching[145]. One of the most known altered pathways is the PI3K/AKT pathway, which can increase glucose uptake and glycolysis in the cells[146]. Higher glycolysis leads to more production of pyruvates, which are often converted to acetyl-CoA for ATP production and synthesis of other macromolecules such as lipids and amino acids. Mitochondria serve as the only source of the acetyl-CoA conversion from pyruvates via its pyruvate dehydrogenase (PDH) complex during the TCA cycle[147]. To transport these acetyl-CoAs to cytoplasm requires an additional step of converting acetyl-CoA to citrate by mitochondrial-specific citrate synthase. Transported citrates are then converted back to acetyl-CoA via ATP-citrate lyase (ACL) as the resources for lipid and protein synthesis[147]. Activated AKT is required for ACL to work, in which AKT phosphorylates ACL for its activation, thus producing more acetyl-CoA in the cytoplasm to fulfill the higher needs of cancer cells[148,149]. This AKT/ACL interaction is supported by the fact that inhibition of ACL enzyme causes a reduction in cell proliferation and tumorigenesis, despite increased glucose uptake[148,149]. Thus, these findings imply that metabolic programming via mitochondrial acetyl-CoA and citrate is the main oncogenic action of PI3K/AKT in cancer cells.
Another known oncogenic action affecting mitochondrial function is the relationship between the cell growth regulators mTOR and hypoxia-inducible factor 1 (HIF-1). mTOR positively influences HIF-1 action during hypoxia[150], and HIF-1 is known to increase glycolytic metabolism[151]. Another effect of HIF-1 is the activation of pyruvate dehydrogenase kinase 1 (PDK1) expression, which inhibits PDH activity in mitochondria[152-154]. This suppression of PDH activity limits the conversion of pyruvates to acetyl-CoA and shifts the conversion of pyruvates to lactate[152-154]. Similarly, MYC oncogenic action is through the mitochondria, in which MYC promotes mitochondrial glutamine metabolism by increasing the expression of glutaminase (GLS), an enzyme that deamidates glutamine to glutamate. Glutamate is needed for nucleic acid and amino acid synthesis, which are vital for cancer cell proliferation. Supporting these findings, cancer cells that express oncogenic MYC, cause growth suppression[155] and prevent the Rho GTPase-induced cancer cell transformation and proliferation[156].
Loss of tumor suppressor expression, p53 for example, can also contribute to mitochondrial energy switching. In healthy cells, p53 suppresses the expression of glucose transporters (GLUT1 and GLUT4) as a mechanism to control glucose uptake by cells[157,158]. p53 protein also suppresses the expression of lactate transporter, monocarboxylic acid transporter 1 (MCT1), to inhibit cellular lactate export, and thus controlling the tumor microenvironment[159]. p53 also controls the rate of glycolysis by activating the expression of TP53-induced glycolysis and apoptosis regulator enzyme (TIGAR)[160] and reducing the expression of the glycolytic enzyme phosphoglycerate mutase (PGM)[161]. Loss of p53 expression causes a reduction of OXPHOS activity with evidence of low mitochondrial complex IV activity[162]. In another study, reduced p53 expression decreased mitochondrial mass and mtDNA copy numbers[163,164]. Importantly, p53 protein is also responsible for inhibiting the oncogenic PI3K/AKT and mTOR pathways[144], thus supporting the notion that the loss of p53 expression can initiate metabolic switching via mitochondrial dysfunction.
Mitochondria and neurodegenerative diseases
The most common clinical manifestations of mitochondrial diseases are neurological and neuromuscular syndromes[165]. There are two theories that can best explain the role of mitochondria in neurodegenerative diseases. First, a decrease in energy production leads to neuronal depolarization that activates the excitatory amino acid receptors and impairs intracellular Ca2+ homeostasis. This situation is followed by protease activation and cell death, which finally leads to neurodegenerative diseases[166]. Second is that mitochondria are the source of ROS via the OXPHOS process, particularly from complex I and III of the ETC. Leakage of electrons from complex I and III produce mitochondrial superoxide, which could cause further damage to macromolecules such as proteins, lipids, and DNA, subsequently leading to a reduced ability of mitochondria to perform their functions. ROS could also activate the apoptosis process via the mitochondrial apoptotic pathway by releasing cytochrome C (Cyto-C) from mitochondria to the cytosol. Pro-apoptotic signals such as Bcl-2 family proteins (Bax and Bak) are translocated into the mitochondria leading to mitochondrial transmembrane permeabilization (MMP)[167]. Active Bax and Bak are inserted into the outer mitochondrial membrane (OMM), resulting in increased MMP[168]. Subsequently, molecules such as Cyto-C, AIF, Smac/DIABLO (second mitochondria-derived activator of caspase/direct inhibitor of apoptosis protein-binding protein with low pI) and Omi/HrtA2 are released into the cytoplasm[169-172]. The formation of the caspase-activating complex or apoptosome composed of Cyto-C, Apaf-1, dATP and procaspase-9, thereby induce the activation of the effector caspases required for apoptosis[173].
Oxidative stress and mitochondrial dysfunction are associated with several neurodegenerative diseases such as Alzheimer’s, Parkinson’s, Friedreich ataxia, and amyotrophic lateral sclerosis (ALS)[174]. The first mtDNA disease was identified in 1988 in a patient with LHON, which is caused by mutations in genes encoding OXPHOS subunit I, III, IV, and V[71]. MTND4*LHON11778 is the most common cause of LHON, representing about 40%-60% of all LHON cases[71]. SOD1 gene mutations cause ALS with increased cellular oxidative stress[175]. SOD1 gene encodes SOD1 protein, which acts as an antioxidant defense mechanism for ROS detoxification. Mitochondrial dysfunction such as defective OXPHOS complexes, changes in mitochondrial membrane potential (MMP), high level of oxidative stress, and decreased mitophagy have been associated with idiopathic Parkinson’s disease (PD)[176]. Antony and colleagues showed that mitochondria of idiopathic PD undergo morphological changes and increased resistance to depolarization. The basal mitochondrial membrane potential (Ψm) of a skin biopsy from an idiopathic PD patient was higher compared to healthy control. The mitochondrial morphology parameters such as node degree, mean volume, skeleton size, perimeter, form factor, node count, erosion body count, endpoints, and mitochondria count were reduced in idiopathic PD compared to control[177]. Previously, research on the pathogenesis of PD has been focusing on oxidative stress, mitochondrial bioenergetics defects, and apoptosis mechanisms. Currently, it has been postulated that the crosstalk dysregulation between mitochondria and endoplasmic reticulum, as well as lysosome, may lead to mitochondrial dysfunction, including a bioenergetics defect, abnormal protein aggregation, and finally neuronal cell death[178].
Multi-omics technologies to diagnose mitochondrial disease
The main problems in diagnosing mitochondrial disease are the heterogeneity of the individuals in their clinical presentation and the incomplete understanding of the disease pathophysiology[1,179]. Advances in molecular technologies, particularly the high-throughput omics that can characterize and identify many targets in a single run[180], have enabled a more in-depth and accurate diagnosis of mitochondrial diseases and their causes. This section intends to summarize the roles of these omic technologies in mitochondrial disease diagnosis.
Genomics
Genomics research for mitochondrial diseases started about 30 years ago when a report of the small sequence of the mitochondrial genome was published[12,13]. Afterwards, various findings of the novel mtDNA mutations associated with diseases were reported, including the early discoveries of mtDNA mutations in ragged-red fiber myopathies[181] and LHON[71]. During this time, it was clear that the mtDNA mutations were not solely the main culprit, but that other nuclear DNA mutations may play a role in the disease, as in some patients, the lack of mtDNA mutations were observed[182]. This situation was evident in OXPHOS deficiency when a report of the succinate dehydrogenase-complex flavoprotein subunit A (SDHA) mutations in Leigh syndrome (a disease caused by the deficiency of oxidative phosphorylation complex proteins) was published, in which the SDHA gene mutations resulted in the deficiency of complex II protein in the patient[183]. Thus, identification of mtDNA mutations and any related gene mutations is important for unraveling the disease-causing or underlying cause of the mitochondrial disease.
In the early days, there was a limitation in detecting the mutations, as most of the disease-causing mutations were identified using the single candidate gene sequencing techniques, and these methods were time- and resource-consuming[184]. The most popular methods were Sanger sequencing and Affymetrix’s DNA re-sequencing MitoChip[184]. To address the issue with heteroplasmy, several other methods were also developed, including PCR-RFLP analysis[50], allele-specific oligonucleotide dot-blot analysis[185], real-time amplification refractory mutation system quantitative PCR[186], and pyrosequencing technique[187]. However, these applications could only detect a single or a few candidate mutations.
The emergence of NGS technologies has rapidly reduced the cost and time spent with a substantial improvement in the detection ability that allows for a wide-scale detection of genome changes[180,184]. The introduction of whole-exome sequencing (WES) and whole-genome sequencing (WGS) technologies increased the mutational detection rate in mitochondrial disease diagnosis. This effect was evidenced by the percentage of disease-causing mutations identified during the pre-NGS era (10%-20%) compared to after NGS era (30%-50% in some cohorts)[188-190]. Typically, there are two workflows for the detection of mtDNA mutations[184,191]: (1) direct analysis (detection of mtDNA sequence from samples that are enriched with mtDNA apart from the cellular DNA; and (2) indirect analysis (the mtDNA mutations are obtained as by-products of the high-throughput sequencing reads).
Direct detection of mtDNA is usually done by adding a technique to purify or isolate the mitochondria before the NGS workflow[184,191], such as ultracentrifugation (a density gradients isolation) or the biochemical or mechanical isolation of the organelles. Another approach is to use specific probes or primers to isolate mtDNA, such as in microarray hybridization and PCR-based enrichment methods[184,192,193]. However, it is important to note that using a primer-based method often results in large overlapping regions, and these regions must be removed before the variant calling analysis. The main advantage of this direct method is the elimination of the DNA regions homologous with mtDNA sequences or those known as nuclear mitochondrial DNAs (NUMTs), which exist in various sizes as clones of genuine mtDNA and can be specific to some populations[194]. Therefore, the findings from the direct mtDNA analysis are usually more reliable.
A typical workflow for the indirect mtDNA analysis is through the by-product annotation of the sequencing reads from the WES and WGS. From these WES or WGS sequencing reads, the annotation process also includes a step to map the reads to the mitochondrial genome. Since the average coverage of this mtDNA fraction sequence is higher than the normally targeted gene regions due to the high copy number of mtDNA per cells, the mapping results in good quality data[195]. Because of this high-quality data and the cost-effectiveness of the NGS technologies, the indirect mtDNA analysis has become a favorite tool for mitochondrial disease diagnosis, due to a simple workflow. However, this indirect mtDNA technique has one problem with false-positive results due to NUMTs[184,191]. The inability to confirm whether the mtDNA reads from the WES or WGS sequences are from the nuclear or mitochondrial genome can cause ambiguity of the findings. The simple method to eliminate this issue is to align the raw reads first to the mitochondrial genome and filter the non-aligned sequences, though some NUMTs do exist in the mtDNA genome database; thus, false heteroplasmy can be introduced[184,191]. New software such as MitoSeek can help to address this issue, where this program can extract the mtDNA mutation and heteroplasmy information from WES data[196]. Furthermore, the existence of the databases such as MSeqDR[197], MITOMAP[198], HmtVar[199], HmtDB 2016[200], Leigh Map[201] and others provide the comprehensive mutation-phenotype relationships to allow the interpretation of the WES and WGS analyses and thereby unravel any novel mutations in the patients. The mitochondrial disease-specific detection kits or panels are already on the market to improve diagnosis[202-205]. By combining the databases and NGS technologies, there is a continuous discovery of many mutations responsible for various mitochondrial diseases.
Transcriptomics
Although genomic NGS techniques are powerful enough to diagnose mitochondrial diseases, the rate of detection for disease-causing mutations are only 25%-50% of cases[180,184,192]. To improve this detection rate, an approach to employ the whole transcriptome sequencing technologies such as the RNA-seq by prioritizing the candidate genes (i.e., those genes that are involved in the oxidative phosphorylation pathways, etc.) to complement the DNA sequencing, can unravel hidden or deep intronic mutations which usually are missed from interpretation og WES and WGS data[206]. An example of this RNA-seq approach was in the primary muscle samples of the genetic myopathy patients, in which the RNA-seq was able to identify disease-causing mutations in 21% of cases[207]. It is important to note that the DNA sequencing technique was unable to detect the mutations in these patients[207]. However, the challenges of using RNA-seq are attributed to the transcriptomic profiling issues, such as the batch effects, and the requirement of robust filtering pipelines to confirm the results[180]. Nevertheless, the fact remains that the RNA-seq technique can detect mutations in the patients who are not detected from WES and WGS sequencing.
Proteomics
mtDNA encodes 13 proteins, including the mitochondrial respiratory chain proteins, ribosomal RNAs and transfer RNAs. The remaining mitochondrial proteins, which include the TCA cycle components, β-oxidation, protein transports, and the other respiratory chain subunits, are from nuclear DNA[208]. Therefore, to characterize the proteome profile of mitochondrial diseases can be very challenging. Up until now, the number of the mammalian mitochondrial proteins discovered is about 1,100 to 1,900, based on the classifications in each database[209-213]. One of the earliest databases for the mitochondrial proteome is MITOP, which was released in 1999[210] and followed by the first comprehensive human mitochondrial proteome database, the MitoProteome Project[211]. Currently, MitoProteome contains about 1,705 genes and 3,625 proteins that are associated with mitochondria[211]. After that, various databases with their analysis tools have been released, including the MitoP2[212], MitoMiner[213], and MitoCarta[209] databases.
An example of the mitochondrial dysfunction study using the proteome analysis is the identification of C17orf89 (NDUFAF8) mutation in Leigh syndrome, in which mass spectrometry (MS) crosslinking interactome analysis was able to show C17orf89/NDUFAF8 as a new candidate for the unresolved cases of isolated complex I deficiency[214]. Another study of proteome profiling of the mitochondrial ribosomes revealed that in the small ribosomal subunit, MRPS34 mutations were responsible for the destabilization of the subunit and impaired monosome assembly in the fibroblasts of Leigh syndrome patients[215]. Importantly, the findings[215] were after WES sequencing in those patients, indicating that proteome profiling could also complement WES sequencing to improve the diagnostic detection of mitochondrial disease.
Metabolomics
Due to limited publications, the potential of metabolomics tools to diagnose mitochondrial disease is uncertain. Lactate and pyruvate have been used as biomarkers for mitochondrial dysfunction, though these biomarkers have low sensitivity and specificity[216]. One example is that the lactate stress test was used in the diagnosis of mitochondrial myopathy. However, the sensitivity of the lactate stress test was 69%, but it can complement the other clinical tests to confirm the diagnosis[217,218]. Advances in technologies allows for the application of mass spectrometry-based metabolomics to profile thousands of small metabolites[180]. In a study of the specific subgroup of the Leigh syndrome patients with mutations in the LRPPRC gene, analysis of the blood and urine metabolites revealed that there were 45 distinct metabolites, including ketones, lipids, kynurenine, lactate, and pyruvates[219]. These findings were important in highlighting the role of metabolomics in unraveling the physiology of mitochondrial disease. However, whether these 45 signature metabolites are specific to the subgroup of Leigh syndrome or applicable to all forms of mitochondrial diseases is unknown. Therefore, further works are needed to confirm these findings, especially in a large cohort, to establish the relationship and diagnostic capacity of the metabolomic approach in mitochondrial disease.
Functional genomics
Following the WES or WGS analysis, the presence of the rare variants or variants of unknown clinical significance (VUS) is challenging to interpret for definitive mitochondrial disease diagnosis[220]. Functional genomics is an area of research of using multiple molecular data to interpret or understanding the impact of a DNA sequence variant on complex biological processes in a cell. Typically, functional genomic utilizes high-throughput data of multiple omics from a single patient or a disease, to evaluate the impact of these genetic variants in transcription and protein translation[220,221]. After that, the validation of these variants is achieved by the functional cell- and tissue-based assays, along with animal models to establish the genotype-phenotypes association as evidence for the pathogenicity.
Typically, the first step of validation is via the confirmation of VUS pathogenicity in the mitochondrial OXPHOS system, which can be done via tissue-based assays (e.g., skeletal muscle biopsy) or cell-based assays (e.g., primary fibroblast culture)[222]. An example is the study of genetic myopathy patients, in which the RNA-seq approach in the primary muscle samples was used to complement the DNA sequencing technology to improve the identification of disease-causing mutations[207]. Importantly, in this study[207], the pathogenicity of these two splice site VUS were confirmed by analyzing the results with the tissue expression database (Genotype-Tissue Expression (GTEx) Consortium[223]). They found that these splice site VUS are observable in the muscles but have very little presence in the cultured dermal fibroblasts[207], thus indicating that a correct sampling tissue type does matter to discover the relevant genetic defects. Since the GTEx database is freely accessible online, for any mitochondrial study that lacked the tissue biopsy samples, a comparative analysis via the proxy tissue sample data could further refine the findings. The confirmation of the protein product expression and changes in the tissue and primary cells, via the biochemical assays and protein assays such as SDS-PAGE or BN-PAGE, are also used to complement the DNA sequencing analysis[222].
Once the pathogenicity is confirmed, most studies performed the additional assays to discover the disease mechanism or molecular effects of the VUS in cell lines or animal models[222]. In most cases, the selected genetic variant is introduced into a cell or animal model via the cell-directed mutagenesis or CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats) technology[220,221]. In a study of patients with mitochondrial respiratory chain complex deficiencies, the comprehensive analyses of genetic screening and fibroblast biochemical analysis together with functional cell line assays were able to identify multiple three novel causative variants, in which all of them were pathogenic based on functional cell-based investigation assays[189]. This comprehensive evidence for the pathogenicity of the genetic variants is important to elucidate the disease mechanism. Thus, most disease-specific genomic databases will have reported evidence of pathogenicity to support the clinical significance of the variants found.
Imaging technologies
Another strategy is to complement the NGS data with magnetic resonance imaging (MRI) of the brain or muscles to confirm the changes in the proteins and structures[224]. Most mitochondrial diseases are heterogeneous in clinical presentation and symptoms, which are often mixed between diseases. Since many of the patients exhibit neurological symptoms, the application of MRI can detect these changes. One example is to use magnetic resonance spectroscopy (MRS) to evaluate brain chemistry for the detection of metabolic and oxidative defects[224]. MRS is a non-invasive in vivo brain imaging to detect biochemical metabolites such as N-acetyl aspartate (NAA), lactate, choline, creatine, and myoinositol[224]. Increased lactate levels are a common feature in mitochondrial disease patients, and such lactate elevation has been observed in the brain[225-227], and muscle[228] of patients. However, the findings in the brain are more consistent compared to the muscle[224]. Furthermore, this lactate elevation was evident in the early stage before any abnormalities or lesion could be detected in the brain of the mitochondrial disease animal model[229], indicating the usefulness of this MRS technique to improve the diagnosis. Interestingly, the phenotypic changes at the brain structures can complement the genetic screening analysis. In Leigh syndrome, the MRI scans of patients with confirmed SURF1 and COX mutations (nuclear mutations) have T2-abnormalities in the brainstem nuclei, whereas the caudate and putamen lesions are seen in patients with mtDNA mutations[230-232]. These findings suggest that MRI scans can confirm the genotype-phenotype changes occurring in mitochondrial disease and thereby improve diagnostic yield.
Challenges in diagnosing mitochondrial diseases
Even though quite significant publications of disease-causing mutations are available, challenges remain on how to improve the diagnosis of these mitochondrial diseases in clinical settings, as the rate of detection for disease-causing mutations is only 25%-50% of cases[180,184,192]. Most of the diagnosis approaches are using the NGS technologies, in which the first step is to use the WES approach, followed by a muscle biopsy if more confirmation is needed for the pathogenicity[180]. Various reasons can explain the failure to detect mtDNA mutations in some patients, such as the existence of the difficult-to-detect mutations, including the recurrent de novo mutations[233], splice site defects, mutations in deep intronic or repeated sequences, and others[180,184,192]. One way to address such limitation is to use trio sequencing of parents and child to allow for accurate detections of these difficult-to-detect mutations, as used by the Deciphering Developmental Disorders Project[234], and the Genomics England 100,000 Genomes (100K) Project[235].
With the problems of heteroplasmic mtDNA mutations, many recommend that sequencing of muscle DNA is needed to complement the WES findings, especially with the low mutant load. Since most patients with mitochondrial diseases are usually carrying a mixture of wild-type and mutated mtDNA (heteroplasmic), their clinical manifestations of the disease also depend on the ratio of the mutated to wild-type mtDNA[236]. Some of these low-frequency heteroplasmy variants can turn into deleterious high heteroplasmy variants[237], and could thereby further complicate the diagnosis. Integrated analysis of the omics can also help to improve the diagnosis, as multiple omic findings could verify the accuracy of the results. An example is a cohort study of adult mitochondrial disease patients with the mtDNA mutation m.3243 A à G, in which the combined analysis of proteomics and metabolomics of their urine samples showed very distinct alterations in lysosomal proteins, calcium-binding proteins, and antioxidant defenses[238]. Importantly, these changes were evident in the asymptomatic carriers of m.3243A>G[238], therefore suggesting the plausibility of a new and early screening strategy of this type of mutation in the patients and their families.
Another issue is the presence of the NUMTs that could interfere with WES or WGS data interpretation and analysis[194]. The indirect method using the WES/WGS data to identify the mitochondrial mutations is a favorable approach due to its cost-effectiveness and high reproducibility. However, the presence of the NUMTs gives some ambiguity to the results[184,191]. Thus, some studies have opted for an addition of the mitochondrial isolation step in the workflow before RNA extraction and sequencing steps to eliminate the NUMTs. However, the resources used are enormous and labor-intensive[191]. Similarly, the proteomic approach for the mitochondrial study also faces the challenges of getting pure mitochondrial proteins[208]. To enrich these mitochondria, many methods have been developed, including the mechanical or chemical disruption method, the differential centrifugation method, and recently introduced magnetic device method[239-241]. However, mitochondrial proteins have dynamic ranges; thus, the samples usually undergo fractionation to reduce their complexity before the analysis[208], which could increase the cost and time spent for each additional procedure. Most studies use sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE), and gel slicing to separate the proteins, followed by high-performance LC-MS analysis[208,239,242-244]. Despite the vast potential of these proteomic applications to diagnose mitochondrial disease, the problems lie within the diversity and tissue-specific expression of these mitochondrial proteins. Currently, only indirect measurements are available to detect them[208]. Moreover, the lack of methods to differentiate between the mitochondrial and cytoplasmic functions of these proteins[208] also contribute to the problems. In addition, there are also the issues of technical expertise to use the proteome interactome analysis tools, and the expensive cost to run the comprehensive proteome profiling[208]. Therefore, innovative approaches and advancement of the proteomic applications in the future are needed to solve these issues, and hopefully to increase the potential of these proteomic applications in diagnosing mitochondrial diseases.
Another improvement for the diagnosis of mitochondrial disease using the genetic data is to perform periodic reanalysis of WES/WGS data of the patients, using various or newly improved bioinformatic pipelines, until the phenotypes changes[245]. In disease with heterogenous genetic and phenotypes involved, the diagnostic yield of using the WES or WGS data ranges from 15% to 50% depending on the options used during the inclusion, selection, and stringency criteria of the analysis. The results also depend on the status of the knowledge and genetic databases on the time of sampling, as this research area is continually expanding[245,246]. Previous studies showed a significant improvement in the diagnostic yield if the patient’s genomic data is re-analyzed again with an improved bioinformatics pipeline[247,248]. Therefore, a periodic re-analysis of the WES or WSG data in one or two years will improve the identification of the disease-causing mutations.
The most important issue is the plausibility and effectiveness of these applications for routine diagnostics of mitochondrial disease. To run all these high-throughput technologies needs high expertise and advanced machine, which are simply not possible for most of the community clinics. Until there is some development on the cutting-edge technologies to deliver the same comprehensive results as the NGS, but with a far less expensive machine, this issue remains a challenge. Another associated problem is the clinical interpretation of the variants or mutations of unknown significant found from the WES or WGS, which can be confusing to the clinicians even with a genetic counseling background. Thus, the existence of the Clinical Genome Resource[249] and ClinVar[250] online databases help in terms of the interpretation of the genomic variants for the clinicians, which are based on the functional characterization of those variants in various disease models. Despite these challenges, NGS tools are already being used to diagnose mitochondrial diseases in many laboratories, as the potential for screening and diagnosis outweigh the downsides of these applications.
Mitochondrial DNA genetic testing available on the market
Several genetic testing approaches are available on the market nowadays. Here, we discuss the availability of genetic testing for mitochondrial diseases. Centogene is one of the rare disease companies that offer testing for 6,500 genes, which is facilitated by highly innovative analytical platforms based on genomics, proteomics, and metabolomics (https://www.centogene.com). Another company, GeneDX, founded in 2000 by two researchers from the National Institute of Health (NIH), USA, was known as a world genomic leader due to their expertise in a rare and ultra-rare genetic disorder analysis. They offer services for the identification of 20,000 genes and hundreds of rare diseases. Also, they offer the chromosomal abnormalities test (https://www.genedx.com). Family Tree DNA is based in Houston, Texas. They provide services for mitochondrial diseases, for example, the autosomal DNA testing, Y DNA testing, and mtDNA testing (http://www.familytreedna.com). Genebase is one of the established direct-to-consumer (DTC) personal genomics company founded in 2005 based in Vancouver, Canada. Genebase received the world’s prestigious DNA laboratory accreditations, such as the American Association of Blood Banks (AABB), College of American Pathology (CAP), Clinical Laboratory Improvement Amendments (CLIA), and ISO17025 for their services (https://www.genebase.com). 23andMe is also a DTC company, and they offer a simple DNA test from saliva, which can be done at home. They provide the test kit for analysis with all the DNA information directly to the customers (https://www.23andme.com). YSEQ company was established in 2013 by Thomas and Astrid Krahn, which offer several platforms for ancestry study and testing, such as Y-SNPs, Y-STRs, and whole-genome testing with an application for rare disease (https://www.yseq.net/). FullGenomes is one of the DTC company founded in 2013 to make NGS technology possible for the public. They offer products such as whole-genome sequencing and interpretation (SNP, STR, and phylogeny) (https://www.fullgenomes.com). Living DNA was founded in 2016 by David and Hannah Nicholson to provide a DTC NGS screening kit (https://livingdna.com). Oxford Ancestors was founded in 2000 by Prof. Bryan Sykes, and they provide the test for mtDNA maternal and yDNA for paternal ancestry (http://www.oxfordancestors.com) [Table 3].
mtDNA tests available on the market
| Company | URL | Year founded, location | Method | Main purpose of test |
|---|---|---|---|---|
| Centogene | https://www.centogene.com | 2006, UK | Whole-exome sequencing | Genealogy |
| GeneDX | https://www.genedx.com | 2000 | ||
| Family Tree DNA | http://www.familytreedna.com | 1999, USA | Next-generation sequencing 2000x coverage | Mitochondrial DNA analysis - limited hypervariable region test or full sequence test |
| Gene Base Systems, Inc | https://www.genebase.com | 2005, Vancouver, British, Columbia, Canada | Advanced fluorescent probe technology for sequencing DNA | Genealogy |
| 23andMe, Inc | https://www.23andme.com | 2006, California | Illumina Global Screening Array (GSA) customized chip | Ancestry, DNA relatives |
| YSEQ | https://www.yseq.net/ | 2013 | Sequencing | Ancestry |
| Full Genomes Corporation | https://www.fullgenomes.com | 2013, USA | Next-generation sequencing | Focusing on the Y chromosome and WGS |
| Living DNA | https://livingdna.com | 2016, England, UK | Next-generation sequencing | Y-DNA, mtDNA haplogroup |
| Oxford Ancestors | http://www.oxfordancestors.com | 2000, England, UK | Next-generation sequencing | mtDNA, Y chromosome |
Conclusion
Mitochondrial diseases are complex and clinically heterogeneous, making early diagnosis difficult. Advances in omics technology, particularly genomics, NGS via targeted mtDNA genome of WES and WGS enable a comprehensive detection strategy with high sensitivity and specificity. Early identification of affected individuals is important for the patient’s outcome, as early treatment will result in a better prognosis. Importantly, with a simple blood sampling, the cascade screening of the family members can be done with the same strategy.
Declarations
Authors’ contributionsWrote the manuscript and revised the manuscript for intellectual content: Sulaiman SA
Wrote a few parts of the manuscript: Mohd Rani Z, Radin FZM
Planned and wrote the manuscript and also revised the manuscript for intellectual content: Abdul Murad NA
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, et al. Mitochondrial diseases. Nat Rev Dis Primers 2016;2:16080.
2. DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol 2013;9:429-44.
3. Duran J, Martinez A, Adler E. Cardiovascular manifestations of mitochondrial disease. Biology 2019;8:34.
5. McCormick EM, Muraresku CC, Falk MJ. Mitochondrial Genomics: a complex field now coming of age. Curr Genet Med Rep 2018;6:52-61.
6. Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab 2012;26:711-23.
7. Tuppen HAL, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta Bioenerg 2010;1797:113-28.
8. Greaves LC, Nooteboom M, Elson JL, Tuppen HAL, Taylor GA, et al. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet 2014;10:e1004620.
9. Castellanos E, Lanning NJ. Phosphorylation of OXPHOS machinery subunits: functional implications in cell biology and disease. Yale J Biol Med 2019;92:523-31.
10. Natarajan V, Chawla R, Mah T, Vivekanandan R, Tan SY, et al. Mitochondrial dysfunction in age-related metabolic disorders. Proteomics 2020;1800404.
11. Nass S, Nass MM. Intramitochondrial fibers with DNA characteristics. II. enzymatic and other hydrolytic treatments. J Cell Biology 1963;19:613-29.
12. Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coulson AR, et al. Sequence and organization of the human mitochondrial genome. Nature 1981;290:457-65.
13. Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 1999;23:147.
14. D’Souza AR, Minczuk M. Mitochondrial transcription and translation: overview. Essays Biochem 2018;62:309-20.
15. Russell O, Turnbull D. Mitochondrial DNA disease-molecular insights and potential routes to a cure. Exp Cell Res 2014;325:38-43.
16. Bayir H, Kagan VE. Bench-to-bedside review: mitochondrial injury, oxidative stress and apoptosis--there is nothing more practical than a good theory. Critical Care (London, England) 2008;12:206.
17. Wallace DC, Brown MD, Lott MT. Mitochondrial DNA variation in human evolution and disease. Gene 1999;238:211-30.
18. Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 2015;77:753-9.
19. Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003;126:1905-12.
20. DiMauro S. Mitochondrial encephalomyopathies--fifty years on: the Robert Wartenberg Lecture. Neurology 2013;81:281-91.
21. Frazier AE, Thorburn DR, Compton AG. Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J Biol Chem 2019;294:5386-95.
22. Lebon S, Chol M, Benit P, Mugnier C, Chretien D, et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J Med Genet 2003;40:896-9.
23. Chinnery PF, Craven L, Mitalipov S, Stewart JB, Herbert M, et al. The challenges of mitochondrial replacement. PLoS genet 2014;10:e1004315.
24. Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. J Clin Invest 2003;111:303-12.
25. Pek NMQ, Phua QH, Ho BX, Pang JKS, Hor JH, et al. Mitochondrial 3243A > G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis 2019;10:802.
26. Kucharczyk R, Dautant A, Gombeau K, Godard F, Tribouillard-Tanvier D, et al. The pathogenic MT-ATP6 m.8851T>C mutation prevents proton movements within the n-side hydrophilic cleft of the membrane domain of ATP synthase. Biochim Biophys Acta Bioenerg 2019;1860:562-72.
27. Blanco-Grau A, Bonaventura-Ibars I, Coll-Cantí J, Melià MJ, Martinez R, et al. Identification and biochemical characterization of the novel mutation m.8839G>C in the mitochondrial ATP6 gene associated with NARP syndrome. Genes Brain Behav 2013;12:812-20.
28. Mordel P, Schaeffer S, Dupas Q, Laville MA, Gérard M, et al. A 2 bp deletion in the mitochondrial ATP 6 gene responsible for the NARP (neuropathy, ataxia, and retinitis pigmentosa) syndrome. Biochem Biophys Res Commun 2017;494:133-7.
29. Rahman S, Blok RB, Dahl HHM, Danks DM, Kirby DM, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996;39:343-51.
30. Alston CL, Veling MT, Heidler J, Taylor LS, Alaimo JT, et al. Pathogenic Bi-allelic Mutations in NDUFAF8 cause leigh syndrome with an isolated complex I deficiency. Am J Hum Genet 2020;106:92-101.
31. Park SY, Kim SH, Lee YM. Molecular diagnosis of myoclonus epilepsy associated with ragged-red fibers syndrome in the absence of ragged red fibers. Front Neurol 2017;8:520.
32. Mackey DA, Oostra RJ, Rosenberg T, Nikoskelainen E, Bronte-Stewart J, et al. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet 1996;59:481-5.
33. Manickam AH, Michael MJ, Ramasamy S. Mitochondrial genetics and therapeutic overview of Leber’s hereditary optic neuropathy. Indian J Ophthalmol 2017;65:1087-92.
34. Emperador S, López-Gallardo E, Hernández-Ainsa C, Habbane M, Montoya J, et al. Ketogenic treatment reduces the percentage of a LHON heteroplasmic mutation and increases mtDNA amount of a LHON homoplasmic mutation. Orphanet J Rare dis 2019;14:150.
35. Remes AM, Peuhkurinen KJ, Herva R, Majamaa K, Hassinen IE. Kearns-Sayre syndrome case presenting a mitochondrial DNA deletion with unusual direct repeats and a rudimentary RNAase mitochondrial ribonucleotide processing target sequence. Genomics 1993;16:256-8.
36. Poulton J, Deadman M, Mark Gardiner R. Duplications of mitochondrial dna in mitochondrial myopathy. Lancet 1989;333:236-40.
37. Shoubridge EA, Johns T, Karpati G. Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy. Hum Mol Genet 1997;6:2239-42.
38. Pineda M, Ormazabal A, López-Gallardo E, Nascimento A, Solano A, et al. Cerebral folate deficiency and leukoencephalopathy caused by a mitochondrial DNA deletion. Ann Neurol 2006;59:394-8.
39. Patel R, Coulter LL, Rimmer J, Parkes M, Chinnery PF, et al. Mitochondrial neurogastrointestinal encephalopathy: a clinicopathological mimic of Crohn’s disease. BMC Gastroenterol 2019;19:11.
40. Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999;283:689-92.
41. González-Vioque E, Torres-Torronteras J, Andreu AL, Martí R. Limited dCTP availability accounts for mitochondrial DNA depletion in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). PLoS genet 2011;7:e1002035.
42. Shaibani A, Shchelochkov OA, Zhang S, Katsonis P, Lichtarge O, et al. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol 2009;66:1028-32.
43. Tang S, Dimberg EL, Milone M, Wong LJC. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol 2012;259:862-8.
44. Saneto RP, Cohen BH, Copeland WC, Naviaux RK. Alpers-Huttenlocher syndrome. Pediatr Neurol 2013;48:167-78.
45. El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 2013;10:186-98.
46. Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes--many genes, common mechanisms. Neuromuscul Disord 2010;20:429-37.
47. Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol 2019;15:40-52.
48. de Vries DD, van Engelen BGM, Gabreëls FJM, Ruitenbeek W, van Oost BA. A second missense mutation in the mitochondrial ATPase 6 gene in Leigh’s syndrome. Ann Neurol 1993;34:410-2.
49. Harding AE, Holt IJ, Sweeney MG, Brockington M, Davis MB. Prenatal diagnosis of mitochondrial DNA8993 T----G disease. Am J Hum Genet 1992;50:629-33.
50. Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet 1990;46:428-33.
51. Carrozzo R, Murray J, Santorelli FM, Capaldi RA. The T9176G mutation of human mtDNA gives a fully assembled but inactive ATP synthase when modeled in Escherichia coli. FEBS Letters 2000;486:297-9.
52. Verny C, Guegen N, Desquiret V, Chevrollier A, Prundean A, et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion 2011;11:70-5.
53. Thyagarajan D, Shanske S, Vazquez-Memije M, Devivo D, Dimauro S. A novel mitochondrial ATPase 6 point mutation in familial bilateral striatal necrosis. Ann Neurol 1995;38:468-72.
54. Moslemi AR, Darin N, Tulinius M, Oldfors A, Holme E. Two new mutations in the MTATP6 gene associated with Leigh syndrome. Neuropediatrics 2005;36:314-8.
55. Tiranti V, Corona P, Greco M, Taanman JW, Carrara F, et al. A novel frameshift mutation of the mtDNA COIII gene leads to impaired assembly of cytochrome c oxidase in a patient affected by Leigh-like syndrome. Hum Mol Genet 2000;9:2733-42.
56. Crimi M, Papadimitriou A, Galbiati S, Palamidou P, Fortunato F, et al. A new mitochondrial DNA mutation in ND3 gene causing severe Leigh syndrome with early lethality. Pediatr Res 2004;55:842-6.
57. McFarland R, Kirby DM, Fowler KJ, Ohtake A, Ryan MT, et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann Neurol 2004;55:58-64.
58. Taylor RW, Singh-Kler R, Hayes CM, Smith PE, Turnbull DM. Progressive mitochondrial disease resulting from a novel missense mutation in the mitochondrial DNA ND3 gene. Ann Neurol 2001;50:104-7.
59. Sarzi E, Brown MD, Lebon S, Chretien D, Munnich A, et al. A novel recurrent mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am J Med Genet A 2007;143a:33-41.
60. Kirby DM, McFarland R, Ohtake A, Dunning C, Ryan MT, et al. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J Med Genet 2004;41:784-9.
61. Deschauer M, Bamberg C, Claus D, Zierz S, Turnbull DM, et al. Late-onset encephalopathy associated with a C11777A mutation of mitochondrial DNA. Neurology 2003;60:1357-9.
62. Komaki H, Akanuma J, Iwata H, Takahashi T, Mashima Y, et al. A novel mtDNA C11777A mutation in Leigh syndrome. Mitochondrion 2003;2:293-304.
63. Taylor RW, Morris AA, Hutchinson M, Turnbull DM. Leigh disease associated with a novel mitochondrial DNA ND5 mutation. Eur J Hum Genet 2002;10:141-4.
64. Simon DK, Friedman J, Breakefield XO, Jankovic J, Brin MF, et al. A heteroplasmic mitochondrial complex I gene mutation in adult-onset dystonia. Neurogenetics 2003;4:199-205.
65. Kirby DM, Kahler SG, Freckmann ML, Reddihough D, Thorburn DR. Leigh disease caused by the mitochondrial DNA G14459A mutation in unrelated families. Ann Neurol 2000;48:102-4.
66. Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci U S A 1994;91:6206-10.
67. Solano A, Roig M, Vives-Bauza C, Hernandez-Pena J, Garcia-Arumi E, et al. Bilateral striatal necrosis associated with a novel mutation in the mitochondrial ND6 gene. Ann Neurol 2003;54:527-30.
68. Ugalde C, Triepels RH, Coenen MJ, van den Heuvel LP, Smeets R, et al. Impaired complex I assembly in a Leigh syndrome patient with a novel missense mutation in the ND6 gene. Ann Neurol 2003;54:665-9.
69. Howell N, Bindoff LA, McCullough DA, Kubacka I, Poulton J, et al. Leber hereditary optic neuropathy: identification of the same mitochondrial ND1 mutation in six pedigrees. Am J Hum Genet 1991;49:939-50.
70. Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet 1991;48:1147-53.
71. Wallace D, Singh G, Lott M, Hodge J, Schurr T, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988;242:1427-30.
72. Brown MD, Voljavec AS, Lott MT, MacDonald I, Wallace DC. Leber’s hereditary optic neuropathy: a model for mitochondrial neurodegenerative diseases. Faseb J 1992;6:2791-9.
73. Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem Biophys Res Commun 1992;187:1551-7.
74. Tawata M, Ohtaka M, Iwase E, Ikegishi Y, Aida K, et al. New mitochondrial DNA homoplasmic mutations associated with Japanese patients with type 2 diabetes. Diabetes 1998;47:276-7.
75. Blakely EL, de Silva R, King A, Schwarzer V, Harrower T, et al. LHON/MELAS overlap syndrome associated with a mitochondrial MTND1 gene mutation. Eur J Hum Genet 2005;13:623-7.
76. Sakuta R, Goto Y, Nonaka I, Horai S. An A-to-G transition at nucleotide pair 11084 in the ND4 gene may be an mtDNA polymorphism. Am J Hum Genet 1993;53:964-5.
77. Lertrit P, Noer AS, Jean-Francois MJ, Kapsa R, Dennett X, et al. A new disease-related mutation for mitochondrial encephalopathy lactic acidosis and strokelike episodes (MELAS) syndrome affects the ND4 subunit of the respiratory complex I. Am J Hum Genet 1992;51:457-68.
78. Liolitsa D, Rahman S, Benton S, Carr LJ, Hanna MG. Is the mitochondrial complex I ND5 gene a hot-spot for MELAS causing mutations? Ann Neurol 2003;53:128-32.
79. Crimi M, Galbiati S, Moroni I, Bordoni A, Perini MP, et al. A missense mutation in the mitochondrial ND5 gene associated with a Leigh-MELAS overlap syndrome. Neurology 2003;60:1857-61.
80. Santorelli FM, Tanji K, Kulikova R, Shanske S, Vilarinho L, et al. Identification of a novel mutation in the mtDNA ND5 gene associated with MELAS. Biochem Biophys Res Commun 1997;238:326-8.
81. Corona P, Antozzi C, Carrara F, D’Incerti L, Lamantea E, et al. A novel mtDNA mutation in the ND5 subunit of complex I in two MELAS patients. Ann Neurol 2001;49:106-10.
82. Ravn K, Wibrand F, Hansen FJ, Horn N, Rosenberg T, et al. An mtDNA mutation, 14453G-->A, in the NADH dehydrogenase subunit 6 associated with severe MELAS syndrome. Eur J Hum Genet 2001;9:805-9.
83. De Coo IF, Renier WO, Ruitenbeek W, Ter Laak HJ, Bakker M, et al. A 4-base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann Neurol 1999;45:130-3.
84. Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990;348:651-3.
85. Varlamov DA, Kudin AP, Vielhaber S, Schroder R, Sassen R, et al. Metabolic consequences of a novel missense mutation of the mtDNA CO I gene. Hum Mol Genet 2002;11:1797-805.
86. Andreu AL, Hanna MG, Reichmann H, Bruno C, Penn AS, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med 1999;341:1037-44.
87. Comi GP, Bordoni A, Salani S, Franceschina L, Sciacco M, et al. Cytochrome c oxidase subunit I microdeletion in a patient with motor neuron disease. Ann Neurol 1998;43:110-6.
88. Clark KM, Taylor RW, Johnson MA, Chinnery PF, Chrzanowska-Lightowlers ZM, et al. An mtDNA mutation in the initiation codon of the cytochrome C oxidase subunit II gene results in lower levels of the protein and a mitochondrial encephalomyopathy. Am J Hum Genet 1999;64:1330-9.
89. Campos Y, Garcia-Redondo A, Fernandez-Moreno MA, Martinez-Pardo M, Goda G, et al. Early-onset multisystem mitochondrial disorder caused by a nonsense mutation in the mitochondrial DNA cytochrome C oxidase II gene. Ann Neurol 2001;50:409-13.
90. Wong LJ, Dai P, Tan D, Lipson M, Grix A, et al. Severe lactic acidosis caused by a novel frame-shift mutation in mitochondrial-encoded cytochrome c oxidase subunit II. Am J Med Genet 2001;102:95-9.
91. Hanna MG, Nelson IP, Rahman S, Lane RJ, Land J, et al. Cytochrome c oxidase deficiency associated with the first stop-codon point mutation in human mtDNA. Am J Hum Genet 1998;63:29-36.
92. Manfredi G, Schon EA, Moraes CT, Bonilla E, Berry GT, et al. A new mutation associated with MELAS is located in a mitochondrial DNA polypeptide-coding gene. Neuromuscul Disord 1995;5:391-8.
93. Temperley RJ, Seneca SH, Tonska K, Bartnik E, Bindoff LA, et al. Investigation of a pathogenic mtDNA microdeletion reveals a translation-dependent deadenylation decay pathway in human mitochondria. Hum Mol Genet 2003;12:2341-8.
94. Wibrand F, Ravn K, Schwartz M, Rosenberg T, Horn N, et al. Multisystem disorder associated with a missense mutation in the mitochondrial cytochrome b gene. Ann Neurol 2001;50:540-3.
95. Schuelke M, Krude H, Finckh B, Mayatepek E, Janssen A, et al. Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation. Ann Neurol 2002;51:388-92.
96. Andreu AL, Bruno C, Dunne TC, Tanji K, Shanske S, et al. A nonsense mutation (G15059A) in the cytochrome b gene in a patient with exercise intolerance and myoglobinuria. Ann Neurol 1999;45:127-30.
97. Legros F, Chatzoglou E, Frachon P, Ogier De Baulny H, Laforet P, et al. Functional characterization of novel mutations in the human cytochrome b gene. Eur J Hum Genet 2001;9:510-8.
98. Keightley JA, Anitori R, Burton MD, Quan F, Buist NR, et al. Mitochondrial encephalomyopathy and complex III deficiency associated with a stop-codon mutation in the cytochrome b gene. Am J Hum Genet 2000;67:1400-10.
99. Tarnopolsky MA, Simon DK, Roy BD, Chorneyko K, Lowther SA, et al. Attenuation of free radical production and paracrystalline inclusions by creatine supplementation in a patient with a novel cytochrome b mutation. Muscle Nerve 2004;29:537-47.
100. Dumoulin R, Sagnol I, Ferlin T, Bozon D, Stepien G, et al. A novel gly290asp mitochondrial cytochrome b mutation linked to a complex III deficiency in progressive exercise intolerance. Mol Cell Probes 1996;10:389-91.
101. Andreu AL, Bruno C, Shanske S, Shtilbans A, Hirano M, et al. Missense mutation in the mtDNA cytochrome b gene in a patient with myopathy. Neurology 1998;51:1444-7.
102. Pulkes T, Liolitsa D, Nelson IP, Hanna MG. Classical mitochondrial phenotypes without mtDNA mutations: the possible role of nuclear genes. Neurology 2003;61:1144-7.
103. Karadimas CL, Greenstein P, Sue CM, Joseph JT, Tanji K, et al. Recurrent myoglobinuria due to a nonsense mutation in the COX I gene of mitochondrial DNA. Neurology 2000;55:644-9.
104. Kollberg G, Moslemi AR, Lindberg C, Holme E, Oldfors A. Mitochondrial myopathy and rhabdomyolysis associated with a novel nonsense mutation in the gene encoding cytochrome c oxidase subunit I. J Neuropathol Exp Neurol 2005;64:123-8.
105. Rahman S, Taanman JW, Cooper JM, Nelson I, Hargreaves I, et al. A missense mutation of cytochrome oxidase subunit II causes defective assembly and myopathy. Am J Hum Genet 1999;65:1030-9.
106. McFarland R, Taylor RW, Chinnery PF, Howell N, Turnbull DM. A novel sporadic mutation in cytochrome c oxidase subunit II as a cause of rhabdomyolysis. Neuromuscul Disord 2004;14:162-6.
107. Kennaway NG, Burton MD, Hall RE, Johnston WSW, Keightley JA, et al. Mitochondrial myopathy and cytochrome c oxidase (COX) deficiency associated with a 15 bp deletion in the gene for COX subunit III. (Abstract 827)” Neurology 1994;44:A335.
108. Musumeci O, Andreu AL, Shanske S, Bresolin N, Comi GP, et al. Intragenic inversion of mtDNA: a new type of pathogenic mutation in a patient with mitochondrial myopathy. Am J hum genet 2000;66:1900-4.
109. Valnot I, Kassis J, Chretien D, de Lonlay P, Parfait B, et al. A mitochondrial cytochrome b mutation but no mutations of nuclearly encoded subunits in ubiquinol cytochrome c reductase (complex III) deficiency. Human genetics 1999;104:460-6.
110. Andreu AL, Checcarelli N, Iwata S, Shanske S, DiMauro S. A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediatr Res 2000;48:311-4.
111. Gattermann N, Retzlaff S, Wang YL, Hofhaus G, Heinisch J, et al. Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia. Blood 1997;90:4961-72.
112. Pandya A, Xia XJ, Erdenetungalag R, Amendola M, Landa B, et al. Heterogenous point mutations in the mitochondrial tRNA Ser(UCN) precursor coexisting with the A1555G mutation in deaf students from Mongolia. Am J Hum Genet 1999;65:1803-6.
113. Wang Q, Li R, Zhao H, Peters JL, Liu Q, et al. Clinical and molecular characterization of a Chinese patient with auditory neuropathy associated with mitochondrial 12S rRNA T1095C mutation. Am J Med Genet A 2005;133A:27-30.
114. Shoffner JM, Brown MD, Torroni A, Lott MT, Cabell MF, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 1993;17:171-84.
115. Kosel S, Egensperger R, Mehraein P, Graeber MB. No association of mutations at nucleotide 5460 of mitochondrial NADH dehydrogenase with Alzheimer’s disease. Biochem Biophys Res Commun 1994;203:745-9.
116. Lin FH, Lin R, Wisniewski HM, Hwang YW, Grundke-Iqbal I, et al. Detection of point mutations in codon 331 of mitochondrial NADH dehydrogenase subunit 2 in Alzheimer’s brains. Biochem Biophys Res Commun 1992;182:238-46.
117. Petruzzella V, Chen X, Schon EA. Is a point mutation in the mitochondrial ND2 gene associated with Alzheimer’s disease. Biochem Biophys Res Commun 1992;186:491-7.
118. Online Mendelian Inheritance in Man O. Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine. Baltimore, MD: Johns Hopkins University; 2019.
119. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010;11:872-84.
120. Geto Z, Molla MD, Challa F, Belay Y, Getahun T. Mitochondrial dynamic dysfunction as a main triggering factor for inflammation associated chronic non-communicable diseases. J Inflamm Res 2020;13:97-107.
121. Niyazov DM, Kahler SG, Frye RE. Primary mitochondrial disease and secondary mitochondrial dysfunction: importance of distinction for diagnosis and treatment. Mol Syndromol 2016;7:122-37.
122. Morava E, van den Heuvel L, Hol F, de Vries MC, Hogeveen M, et al. Mitochondrial disease criteria: Diagnostic applications in children. Neurology 2006;67:1823-6.
123. Bris C, Goudenege D, Desquiret-Dumas V, Charif M, Colin E, et al. Bioinformatics Tools and Databases to Assess the Pathogenicity of Mitochondrial DNA Variants in the Field of Next Generation Sequencing. Front Genet 2018;9:632.
124. Xu J, Kitada M, Koya D. The impact of mitochondrial quality control by Sirtuins on the treatment of type 2 diabetes and diabetic kidney disease. Biochim Biophys Acta Mol Basis Dis 2020;1866:165756.
125. Li K, Wu L, Liu J, Lin W, Qi Q, et al. Maternally inherited diabetes mellitus associated with a Novel m.15897G>A mutation in mitochondrial tRNA(Thr) gene. J Diabetes Res 2020;2020:2057187.
126. Sangwung P, Petersen KF, Shulman GI, Knowles JW. Mitochondrial dysfunction, insulin resistance and potential genetic implications. Endocrinology 2020; doi: 10.1210/endocr/bqaa017.
127. Pereira CA, Carlos D, Ferreira NS, Silva JF, Zanotto CZ, et al. Mitochondrial DNA promotes NLRP3 inflammasome activation and contributes to endothelial dysfunction and inflammation in Type 1 diabetes. Front Physiol 2020;10:1557.
128. Docrat TF, Nagiah S, Naicker N, Baijnath S, Singh S, et al. The protective effect of metformin on mitochondrial dysfunction and endoplasmic reticulum stress in diabetic mice brain. Eur J Pharmacol 2020;875:173059.
129. Umbria M, Ramos A, Aluja MP, Santos C. The role of control region mitochondrial DNA mutations in cardiovascular disease: stroke and myocardial infarction. Sci Rep 2020;10:2766.
130. Hirata Y, Inoue A, Suzuki S, Takahashi M, Matsui R, et al. Trans-Fatty acids facilitate DNA damage-induced apoptosis through the mitochondrial JNK-Sab-ROS positive feedback loop. Sci Rep 2020;10:2743.
131. Aguilar-López BA, Moreno-Altamirano MMB, Dockrell HM, Duchen MR, Sánchez-García FJ. Mitochondria: an integrative Hub coordinating circadian rhythms, metabolism, the microbiome, and immunity. Front Cell Dev Biol 2020;8:51.
132. Yang S, Liu Y, Guo Y, Liu R, Qi F, et al. Circadian gene clock participates in mitochondrial apoptosis pathways by regulating mitochondrial membrane potential, mitochondria out membrane permeablization and apoptosis factors in AML12 hepatocytes. Mol Cell Biochem 2020; doi: 10.1007/s11010-020-03701-1.
133. Sardon Puig L, Valera-Alberni M, Cantó C, Pillon NJ. Circadian rhythms and mitochondria: connecting the dots. Front genet 2018;9:452.
134. Rahman A, Hasan AU, Nishiyama A, Kobori H. Altered circadian timing system-mediated non-dipping pattern of blood pressure and associated cardiovascular disorders in metabolic and kidney diseases. Int J Mol Sci 2018;19:400.
135. Gombert M, Carrasco-Luna J, Pin-Arboledas G, Codoñer-Franch P. The connection of circadian rhythm to inflammatory bowel disease. Trans Res 2019;206:107-18.
136. Maiese K. Moving to the rhythm with clock (Circadian) genes, autophagy, mTOR, and SIRT1 in degenerative disease and cancer. Curr Neurovasc Res 2017;14:299-304.
137. Leng Y, Musiek ES, Hu K, Cappuccio FP, Yaffe K. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol 2019;18:307-18.
138. Takaguri A, Sasano J, Akihiro O, Satoh K. The role of circadian clock gene BMAL1 in vascular proliferation. Eur J Pharmacol 2020;872:172924.
139. Škrlec I, Milić J, Steiner R. The impact of the circadian genes CLOCK and ARNTL on myocardial infarction. J Clin Med 2020;9:484.
140. Zhang ZQ, Ding JW, Wang XA, Luo CY, Yu B, et al. Abnormal circadian rhythms are associated with plaque instability in acute coronary syndrome patients. Int J Clin Exp Pathol 2019;12:3761-71.
141. Wang XB, Cui NH, Liu Xn, Liu X. Mitochondrial 8-hydroxy-2’-deoxyguanosine and coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc Diabetol 2020;19:22.
142. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012;21:297-308.
143. Ganapathy-Kanniappan S. Molecular intricacies of aerobic glycolysis in cancer: current insights into the classic metabolic phenotype. Crit Rev Biochem Mol Biol 2018;53:667-82.
144. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, et al. The regulation of AMPK β1, TSC2, and PTEN Expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res 2007;67:3043-53.
145. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010;330:1340-4.
146. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 2004;64:3892-9.
147. Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71:2577-604.
148. Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005;24:6314-22.
149. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005;8:311-21.
150. Land SC, Tee AR. Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biolog Chem 2007;282:20534-43.
151. Sakamoto T, Weng JS, Hara T, Yoshino S, Kozuka-Hata H, et al. Hypoxia-inducible factor 1 regulation through cross talk between mTOR and MT1-MMP. Mol Cell Biol 2014;34:30-42.
152. Kietzmann T, Mennerich D, Dimova EY. Hypoxia-inducible factors (HIFs) and Phosphorylation: impact on stability, localization, and transactivity. Front Cell Dev Biol 2016;4.
153. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006;3:187-97.
154. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177-85.
155. Le A, Lane AN, Hamaker M, Bose S, Gouw A, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012;15:110-21.
156. Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer cell 2010;18:207-19.
157. Itahana Y, Itahana K. Emerging roles of p53 family members in glucose metabolism. Int J Mol Sci 2018;19:776.
158. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res 2004;64:2627-33.
159. Boidot R, Végran F, Meulle A, Le Breton A, Dessy C, et al. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res 2012;72:939-48.
160. Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006;126:107-20.
161. Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, et al. Glycolytic enzymes can modulate cellular life Span. Cancer Res 2005;65:177-85.
162. Zhou S, Kachhap S, Singh KK. Mitochondrial impairment in p53-deficient human cancer cells. Mutagenesis 2003;18:287-92.
163. Lebedeva MA, Eaton JS, Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta 2009;1787:328-34.
164. Kulawiec M, Ayyasamy V, Singh KK. p53 regulates mtDNA copy number and mitocheckpoint pathway. J carcinog 2009;8:8.
165. D’Souza AD, Parikh N, Kaech SM, Shadel GS. Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion 2007;7:374-85.
166. Di Donato S. Disorders related to mitochondrial membranes: pathology of the respiratory chain and neurodegeneration. J Inherit Metab Dis 2000;23:247-63.
167. Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem 2004;90:1281-9.
168. Rehm M, Düßmann H, Jänicke RU, Tavaré JM, Kögel D, et al. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process: role of caspase-3. J Biol Chem 2002;277:24506-14.
169. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000;102:43-53.
170. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996;86:147-57.
171. Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999;397:441-6.
172. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000;102:33-42.
173. Zou H, Li Y, Liu X, Wang X. An APAF-1·Cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biolog Chem 1999;274:11549-56.
174. Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007;145:1233-48.
175. Saccon RA, Bunton-Stasyshyn RKA, Fisher EMC, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain: a journal of neurology 2013;136:2342-58.
176. Grünewald A, Kumar KR, Sue CM. New insights into the complex role of mitochondria in Parkinson’s disease. Prog Neurobiol 2019;177:73-93.
177. Antony PMA, Kondratyeva O, Mommaerts K, Ostaszewski M, Sokolowska K, et al. Fibroblast mitochondria in idiopathic Parkinson’s disease display morphological changes and enhanced resistance to depolarization. Sci Rep 2020;10:1569.
178. Sironi L, Restelli LM, Tolnay M, Neutzner A, Frank S. Dysregulated Interorganellar Crosstalk Of Mitochondria In The Pathogenesis of Parkinson’s disease. Cells 2020;9:233.
179. Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol 2018;19:77-92.
181. Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988;331:717-9.
182. Ingman M, Gyllensten U. mtDB: Human mitochondrial genome database, a resource for population genetics and medical sciences. Nucleic Acids Res 2006;34:D749-51.
183. Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet 1995;11:144-9.
184. Ye F, Samuels DC, Clark T, Guo Y. High-throughput sequencing in mitochondrial DNA research. Mitochondrion 2014;17:157-63.
185. Liang MH, Johnson DR, Wong LJC. Preparation and validation of PCR-generated positive controls for diagnostic dot blotting. Clin Chem 1998;44:1578-9.
186. Bai RK, Wong LJC. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin Chem 2004;50:996-1001.
187. White HE, Durston VJ, Seller A, Fratter C, Harvey JF, et al. Accurate detection and quantitation of heteroplasmic mitochondrial point mutations by pyrosequencing. Genet Test 2005;9:190-9.
188. Pronicka E, Piekutowska-Abramczuk D, Ciara E, Trubicka J, Rokicki D, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med 2016;14:174.
189. Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet 2016;12:e1005679.
190. Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 2012;4:118ra10.
191. Gould MP, Bosworth CM, McMahon S, Grandhi S, Grimberg BT, et al. PCR-free enrichment of mitochondrial DNA from human blood and cell lines for high quality next-generation DNA sequencing. PLoS One 2015;10:e0139253.
192. Vasta V, Ng SB, Turner EH, Shendure J, Hahn SH. Next generation sequence analysis for mitochondrial disorders. Genome Med 2009;1:100.
193. Sosa MX, Sivakumar IKA, Maragh S, Veeramachaneni V, Hariharan R, et al. Next-generation sequencing of human mitochondrial reference genomes uncovers high heteroplasmy frequency. PLoS Comput Biol 2012;8:e1002737.
194. Dayama G, Emery SB, Kidd JM, Mills RE. The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res 2014;42:12640-9.
195. Picardi E, Pesole G. Mitochondrial genomes gleaned from human whole-exome sequencing. Nat Methods 2012;9:523-4.
196. Guo Y, Li J, Li CI, Shyr Y, Samuels DC. MitoSeek: extracting mitochondria information and performing high-throughput mitochondria sequencing analysis. Bioinformatics 2013;29:1210-1.
197. Falk MJ, Shen L, Gonzalez M, Leipzig J, Lott MT, et al. Mitochondrial disease sequence data resource (MSeqDR): a global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol Genet Metabol 2015;114:388-96.
198. Kogelnik AM, Lott MT, Brown MD, Navathe SB, Wallace DC. MITOMAP: a human mitochondrial genome database. Nucleic acids Res 1996;24:177-9.
199. Preste R, Vitale O, Clima R, Gasparre G, Attimonelli M. HmtVar: a new resource for human mitochondrial variations and pathogenicity data. Nucleic Acids Res 2019;47:D1202-10.
200. Clima R, Preste R, Calabrese C, Diroma MA, Santorsola M, et al. HmtDB 2016: data update, a better performing query system and human mitochondrial DNA haplogroup predictor. Nucleic Acids Res 2017;45:D698-706.
201. Rahman J, Noronha A, Thiele I, Rahman S. Leigh map: a novel computational diagnostic resource for mitochondrial disease. Ann Neurol 2017;81:9-16.
202. Abicht A, Scharf F, Kleinle S, Schön U, Holinski-Feder E, et al. Mitochondrial and nuclear disease panel (Mito-aND-Panel): combined sequencing of mitochondrial and nuclear DNA by a cost-effective and sensitive NGS-based method. Mol Genet Genomic Med 2018;6:1188-98.
203. Dames S, Chou LS, Xiao Y, Wayman T, Stocks J, et al. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J Mol Diagnos 2013;15:526-34.
204. Pereira V, Longobardi A, Borsting C. Sequencing of mitochondrial genomes using the precision ID mtDNA whole genome panel. Electrophoresis 2018;39:2766-75.
205. Marquis J, Lefebvre G, Kourmpetis YAI, Kassam M, Ronga F, et al. MitoRS, a method for high throughput, sensitive, and accurate detection of mitochondrial DNA heteroplasmy. BMC Genomics 2017;18:326.
206. Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun 2017;8:15824.
207. Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 2017;9:eaal5209.
208. Gómez-Serrano M, Camafeita E, Loureiro M, Peral B. Mitoproteomics: tackling mitochondrial dysfunction in human disease. Oxid Med Cell Longev 2018;2018:1435934.
209. Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 2016;44:D1251-7.
210. Scharfe C, Zaccaria P, Hoertnagel K, Jaksch M, Klopstock T, et al. MITOP: database for mitochondria-related proteins, genes and diseases. Nucleic Acids Res 1999;27:153-5.
211. Cotter D, Guda P, Fahy E, Subramaniam S. MitoProteome: mitochondrial protein sequence database and annotation system. Nucleic Acids Res 2004;32:D463-7.
212. Elstner M, Andreoli C, Ahting U, Tetko I, Klopstock T, et al. MitoP2: an integrative tool for the analysis of the mitochondrial proteome. Mol Biotechnol 2008;40:306-15.
213. Smith AC, Robinson AJ. MitoMiner, an integrated database for the storage and analysis of mitochondrial proteomics data. Mol Cell Proteomics 2009;8:1324-37.
214. Floyd BJ, Wilkerson EM, Veling MT, Minogue CE, Xia C, et al. Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol Cell 2016;63:621-32.
215. Lake NJ, Webb BD, Stroud DA, Richman TR, Ruzzenente B, et al. Biallelic mutations in MRPS34 lead to instability of the small mitoribosomal subunit and leigh syndrome. Am J Hum Genet 2017;101:239-54.
216. Debray FG, Mitchell GA, Allard P, Robinson BH, Hanley JA, et al. Diagnostic accuracy of blood lactate-to-pyruvate molar ratio in the differential diagnosis of congenital lactic acidosis. Clin Chem 2007;53:916-21.
217. Finsterer J, Eichberger H, Jarius C, Boltzmann L. Lactate-stress testing in 54 patients with mitochondriopathy. Eur Arch Psychiatry Clin Neurosci 2000;250:36-9.
218. Finsterer J, Milvay E. Stress lactate in mitochondrial myopathy under constant, unadjusted workload. Eur J Neurol 2004;11:811-6.
219. Thompson Legault J, Strittmatter L, Tardif J, Sharma R, Tremblay-Vaillancourt V, et al. A metabolic signature of mitochondrial dysfunction revealed through a monogenic form of leigh syndrome. Cell Rep 2015;13:981-9.
220. Rodenburg RJ. The functional genomics laboratory: functional validation of genetic variants. J Inherit Metab Dis 2018;41:297-307.
221. Gasperskaja E, Kučinskas V. The most common technologies and tools for functional genome analysis. Acta Med Litu 2017;24:1-11.
222. Thompson K, Collier JJ, Glasgow RIC, Robertson FM, Pyle A, et al. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J Inherit Metab Dis 2020;43:36-50.
223. Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648-60.
225. Lunsing RJ, Strating K, de Koning TJ, Sijens PE. Diagnostic value of MRS-quantified brain tissue lactate level in identifying children with mitochondrial disorders. Eur Radiol 2017;27:976-84.
226. Chi CS, Lee HF, Tsai CR, Chen WS, Tung JN, et al. Lactate peak on brain MRS in children with syndromic mitochondrial diseases. J Chin Med Assoc 2011;74:305-9.
227. Dinopoulos A, Cecil KM, Schapiro MB, Papadimitriou A, Hadjigeorgiou GM, et al. Brain MRI and proton MRS findings in infants and children with respiratory chain defects. Neuropediatrics 2005;36:290-301.
228. Matthews PM, Allaire C, Shoubridge EA, Karpati G, Carpenter S, et al. In vivo muscle magnetic resonance spectroscopy in the clinical investigation of mitochondria1 disease. Neurology 1991;41:114.
229. Saito S, Takahashi Y, Ohki A, Shintani Y, Higuchi T. Early detection of elevated lactate levels in a mitochondrial disease model using chemical exchange saturation transfer (CEST) and magnetic resonance spectroscopy (MRS) at 7T-MRI. Radiol Phys Technol 2019;12:46-54.
230. Farina L, Chiapparini L, Uziel G, Bugiani M, Zeviani M, et al. MR findings in Leigh syndrome with COX deficiency and SURF-1 mutations. Am J neuroradiol 2002;23:1095-100.
231. Bluml S, Seymour KJ, Ross BD. Developmental changes in choline- and ethanolamine-containing compounds measured with proton-decoupled (31)P MRS in in vivo human brain. Magn Reson Med 1999;42:643-54.
232. Pouwels PJ, Brockmann K, Kruse B, Wilken B, Wick M, et al. Regional age dependence of human brain metabolites from infancy to adulthood as detected by quantitative localized proton MRS. Pediatric Res 1999;46:474-85.
233. Thompson K, Majd H, Dallabona C, Reinson K, King MS, et al. Recurrent de novo dominant mutations in SLC25A4 cause severe early-onset mitochondrial disease and loss of mitochondrial DNA copy number. Am J Hum Genet 2016;99:860-76.
234. Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015;519:223-8.
235. Griffin BH, Chitty LS, Bitner-Glindzicz M. The 100 000 genomes project: what it means for paediatrics. Arch Dis Child Educ Pract Ed 2017;102:105-7.
236. Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 2015;16:530-42.
237. Perales-Clemente E, Cook AN, Evans JM, Roellinger S, Secreto F, et al. Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J 2016;35:1979-90.
238. Hall AM, Vilasi A, Garcia-Perez I, Lapsley M, Alston CL, et al. The urinary proteome and metabonome differ from normal in adults with mitochondrial disease. Kidney Int 2015;87:610-22.
239. Wettmarshausen J, Perocchi F. Isolation of functional mitochondria from cultured cells and mouse tissues. Mitochondria: Practical Protocols. New York: Springer New York; 2017. pp. 15-32.
240. Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat Protoc 2007;2:287-95.
241. Tang B, Zhao L, Liang R, Zhang Y, Wang L. Magnetic nanoparticles: an improved method for mitochondrial isolation. Mol Med Rep 2012;5:1271-6.
242. Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008;134:112-23.
243. Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003;115:629-40.
244. Foster LJ, de Hoog CL, Zhang Y, Zhang Y, Xie X, et al. A mammalian organelle map by protein correlation profiling. Cell 2006;125:187-99.
245. Costain G, Jobling R, Walker S, Reuter MS, Snell M, et al. Periodic reanalysis of whole-genome sequencing data enhances the diagnostic advantage over standard clinical genetic testing. Eur J Hum Genet 2018;26:740-4.
246. Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502-11.
247. Sun Y, Xiang J, Liu Y, Chen S, Yu J, et al. Increased diagnostic yield by reanalysis of data from a hearing loss gene panel. BMC Med Genom 2019;12:76.
248. Salfati EL, Spencer EG, Topol SE, Muse ED, Rueda M, et al. Re-analysis of whole-exome sequencing data uncovers novel diagnostic variants and improves molecular diagnostic yields for sudden death and idiopathic diseases. Genome Med 2019;11:83.
249. Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, et al. ClinGen--the clinical genome resource. N Engl J Med 2015;372:2235-42.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sulaiman SA, Mohd Rani Z, Mohd Radin FZ, Abdul Murad NA. Advancement in the diagnosis of mitochondrial diseases. J Transl Genet Genom 2020;4:159-87. http://dx.doi.org/10.20517/jtgg.2020.27
AMA Style
Sulaiman SA, Mohd Rani Z, Mohd Radin FZ, Abdul Murad NA. Advancement in the diagnosis of mitochondrial diseases. Journal of Translational Genetics and Genomics. 2020; 4(3): 159-87. http://dx.doi.org/10.20517/jtgg.2020.27
Chicago/Turabian Style
Sulaiman, Siti Aishah, Zamzureena Mohd Rani, Fara Zela Mohd Radin, Nor Azian Abdul Murad. 2020. "Advancement in the diagnosis of mitochondrial diseases" Journal of Translational Genetics and Genomics. 4, no.3: 159-87. http://dx.doi.org/10.20517/jtgg.2020.27
ACS Style
Sulaiman, SA.; Mohd Rani Z.; Mohd Radin FZ.; Abdul Murad NA. Advancement in the diagnosis of mitochondrial diseases. J. Transl. Genet. Genom. 2020, 4, 159-87. http://dx.doi.org/10.20517/jtgg.2020.27
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 49 clicks
Cite This Article 49 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.