
New diagnostic pathways for mitochondrial disease

Abstract
Mitochondrial diseases collectively represent the most common cause of inherited metabolic disease. They are estimated to affect at least 1 in 8,000 adults and at least 1 in 250 adults carry a disease-causing genetic mutation. They comprise a heterogeneous group of disorders caused by mutations in either the nuclear or mitochondrial genome, which ultimately result in dysfunction of the critical cellular energy producing mitochondrial respiratory chain. Owing to the key role of mitochondria in energy production, mitochondrial disorders predominantly manifest in tissues with high metabolic demand. However, they demonstrate significant phenotypic and genotypic variability, often rendering the diagnostic process protracted and challenging. Since Luft’s first description of mitochondrial disease nearly 60 years ago, substantial evolution in diagnostic techniques have simultaneously improved the diagnosis and understanding of mitochondrial disease and biology, but the standard diagnostic approach has failed to evolve at the same pace. Although sequencing technologies and analysis for the diagnosis of mitochondrial disease continue to evolve, advances to date, our expanding understanding of mitochondrial diseases and the increasing affordability of these new technologies justify a paradigm shift in the diagnostic approach. We review the progression, impact and challenges of diagnosing mitochondrial diseases and propose a minimally invasive “genetics first” approach incorporating stratification using non-invasive biomarkers, followed by non-targeted next-generation sequencing, such as whole genome sequencing. Such an approach may improve diagnostic yield and streamline diagnosis, leaving invasive investigations to address diagnostic challenges and functional validation of novel variants.
Keywords
Introduction
Mitochondrial diseases comprise a diverse group of genetic disorders characterised by disrupted cellular energy metabolism, which may arise due to mutations in either the mitochondrial (mtDNA) or nuclear (nDNA) genome[1-4]. Collectively, mitochondrial diseases represent the most common cause of inherited metabolic disease[1], estimated to affect at least 1 in 8000 adults[5]. However, population-based studies indicate that the prevalence of mitochondrial disease may be as high as 1 in 250 adults, with the majority of cases being under-recognised[6-8].
Present in all nucleated cells of the body, mitochondria are dynamic intracellular organelles that are central to cellular homeostasis and metabolism. They host a variety of biochemical pathways and play a primary role in energy generation[9]. Consequently, mitochondrial diseases frequently manifest in tissues with high energy requirements[10]. Although mitochondrial diseases may present with one of many well-defined clinical syndromes, clinical manifestations are protean, ranging from single organ, mild or oligo-asymptomatic disease to severe or life-threatening multi-organ dysfunction. Moreover, symptoms and signs may overlap with more common conditions and evolve throughout an individual’s lifespan[4,11-13]. Even for experienced clinicians, the vast clinical and genetic variability can render specific genetic diagnoses challenging, and the process may become a protracted “odyssey”, taking years before achieving molecular diagnosis[14].
Mitochondrial medicine has seen substantial advances in diagnostic technologies over the last 50 years, from the pre-molecular era of histological analysis of muscle to rapidly accelerating identification of the molecular aetiologies of disease using next-generation sequencing (NGS) technologies. The notoriously heterogeneous nature of mitochondrial diseases, their individual rarity, genotypic and phenotypic variability and overlapping presentations with other genetic disorders, make them an ideal candidate group for a non-targeted approach to genetic diagnosis. Although there remain important challenges to such an approach, including optimising bioinformatic pipelines, classification and functional validation of variants and cost, early studies support their utility[3,4,15-23]. Whole exome sequencing (WES) approaches have markedly improved diagnostic yield, highlighted the genetic variability of diseases, facilitated the diagnosis of monogenic mitochondrial mimics and advanced the understanding of mitochondrial biology, opening up potential therapeutic avenues[3,4,15-23]. Whole genome sequencing (WGS) offers further potential, through unbiased, simultaneous bigenomic sequencing with improved coverage, incorporation of non-coding regions and excellent mtDNA coverage depth[24-26].
The traditional and prevailing diagnostic approach, however, recapitulates the technological evolution in mitochondrial disease diagnosis, moving from clinical evaluation to invasive biopsy and subsequently, targeted sequencing, reserving WES or WGS for consideration in undiagnosed cases[4]. In this article, we briefly review the history of mitochondrial disease diagnosis, its evolution, impact and outstanding challenges, and propose an alternative, minimally invasive “genetics first” approach, which complements clinical evaluation with serum biomarkers for stratification, followed by exploratory bigenomic NGS. Recourse to more invasive techniques, including muscle biopsy, is reserved for aetiological uncertainty, identification of tissue-specific variants and functional validation of novel variants. Such an approach has the potential to streamline diagnosis and limit invasive investigations, without increasing costs, whilst optimising reciprocal gains in the understanding of mitochondrial biology and potential therapeutic avenues.
An historical perspective on the approach to diagnosis
Techniques for diagnosing mitochondrial disease have significantly advanced since Ernster and colleagues described the enzyme activity of skeletal muscle mitochondria in 1959[27], paving the way for their identification of Luft’s first reported case of mitochondrial disease[28]. Subsequent development of the modified Gomori trichrome stain[29] allowed rapid identification of “ragged red” fibres on muscle biopsy, the first pathologic hallmark of mitochondrial disease. Clinical-histological descriptions of mitochondrial diseases ensued, and early diagnostic criteria were based on recognising a constellation of features comprising a clinical syndrome - such as mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) - combined with biochemical and/or histopathological evidence from muscle tissues[30-36]. This approach resulted in biases towards identified disease syndromes, and led to underdiagnosis of those with non-classical symptoms[37].
The mitochondrial genome was sequenced in its entirety in 1981[38] and the first two reports of genetic causes of mitochondrial diseases were published in 1988[39,40]. Shortly afterward, the m.3243A>G mutation was identified as the (most common) cause for the MELAS syndrome[41]. Since this discovery, more than 300 pathogenic mtDNA point mutations, deletions and rearrangements have been reported, involving almost all 37 mtDNA-encoded genes[1,42]. Although the nuclear genome encodes a vastly greater proportion of the mitochondrial proteome (~1200 genes)[43], including over 320 genes implicated in disease to date[1,3,4,44-46], causative nuclear gene involvement was only definitively established some years after the first mtDNA mutations were identified[47,48]. Nuclear disease plays a significantly greater role than initially appreciated, accounting for the majority of childhood-onset disease[16] and a substantial proportion of adult-onset mitochondrial disease[5]. Early tools for genetic diagnosis were limited, testing one or a small panel of common mtDNA point mutations, with poor sensitivity for heteroplasmy below ~30%-50% by Sanger sequencing[42]. Despite limitations, the advent of genetic diagnosis permitted greater appreciation of the broad spectrum and phenotypic variability associated with specific mutations and mitochondrial diseases in general[49-52], which has continued to expand alongside improving sequencing techniques.
Technology to facilitate routine clinical genetic diagnosis did not become readily available until the mid-2000s[42] and, historically, relied on sequential Sanger sequencing of clinically prioritised individual genes - a costly, laborious and limited approach, which necessarily biased towards known genotype-phenotype correlations. Consequently, definitive genetic diagnosis was difficult to achieve and molecular diagnosis rates remained low[53], with clinical and biochemical characterisation of greatest utility (albeit imperfect) in confirming or excluding mitochondrial disease. This embedded a “biopsy first” approach, with the clinical and biochemical phenotype guiding targeted genetic testing[4,54]. The advent of powerful, high-throughput, NGS technologies enabling simultaneous interrogation of many, or all genes[55], has transformed genetic diagnosis. Consequently, the genetic landscape of mitochondrial diseases - and understanding of mitochondrial biology - has expanded rapidly over the last two decades, challenging established aetiological concepts of disease and clinical diagnostic approaches.
Limitations of the traditional function to gene approach
Numerous iterations of a diagnostic algorithm for mitochondrial diseases have been proposed. Although the complexity of the disease has precluded consensus, common is a “function to gene” approach centred on muscle biopsy: combining clinical features with biochemical and enzymatic characterisation from muscle biopsy to guide targeted genetic testing. However, there are significant limitations of this approach, prompting calls for a paradigm shift to a “genetics first” approach followed by functional validation.[54,56,57]Figure 1 provides a comparative summary of these approaches.
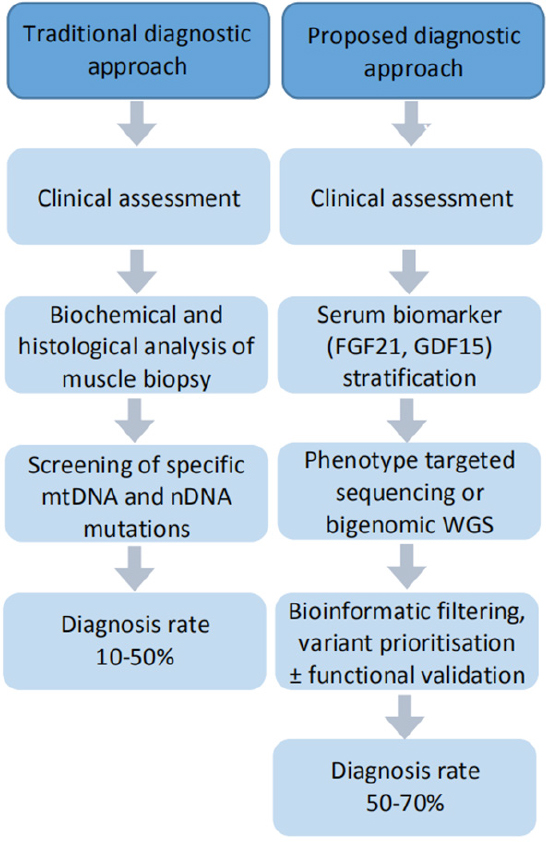
Figure 1. Comparative summary of traditional and proposed diagnostic approaches to mitochondrial diseases. Adapted from Liang et al.[13]
Muscle biopsy can be a helpful diagnostic tool, demonstrating histological and - often more sensitive - ultrastructural changes, as well as providing biochemical and enzymatic information. Muscle tissue may also be utilised for genetic analysis, particularly mtDNA rearrangements, deletion and depletion studies where sensitivity in blood is limited due to heteroplasmy[13,58,59]. However, technological improvements have increased the sensitivity for detecting point mutations and certain deletions in blood or urine[60].
Biopsy is an invasive procedure, which often requires general anaesthetic, presenting significant risk to those with mitochondrial disease, and adding significant cost to the diagnostic process. Furthermore, appropriate preparation, analysis and interpretation of muscle biopsy presents many technical challenges that can impact upon results and in turn, diagnosis and care[61]. Specimen handling, transport and the varied preparation requirements are therefore critical for reliable and reproducible findings, as is interpretation by a clinician with appropriate expertise[13,37,58,62,63]. Biochemical and enzymatic analysis are best performed at an experienced laboratory with established normal criteria. As assays can vary between laboratories, inter-laboratory comparison is challenging[58,62,64,65]. Both false positive and false negative results occur; light microscopy can be normal in up to half of affected patients and findings may evolve over time[37]. Sensitivity is limited, especially for ragged red fibres, which are age-dependent, and in young children and young adults with mitochondrial disease. Specificity is also limited, with mitochondrial changes occurring in a variety of other myopathies including toxic exposures[13,63,66,67]. Ultrastructural changes identified on electron microscopy, although present in up to a third of patients in whom light microscopy is normal, may also be absent in affected patients, and can similarly be seen in various other conditions[37,63], whilst normal respiratory chain biochemical activity in muscle does not exclude mitochondrial disease[68,69]. Although microscopic and biochemical findings from biopsy may provide strong evidence for mitochondrial disease when present, they cannot always differentiate between primary mitochondrial disease and secondary mitochondrial dysfunction, have limited utility to guide specific management and prognosis, and cannot inform genetic counselling[54,58,63].
Clinical characterisation remains imperative - not least to identify and proactively manage organ involvement - with certain phenotypes indicative of a specific or restricted genotype, particularly amongst “classical” mtDNA-based syndromes, such as MELAS. However, even where a defined syndrome is present, genetic heterogeneity is common (e.g., Leigh Syndrome[52]). More often, clinical features do not neatly fit a specific clinical syndrome and presentations can be heterogeneous, with poor phenotype-genotype correlation and therefore, low predictive value for specific genetic diagnosis[61,70]. Muscle biopsy findings do not reliably predict specific genetic aetiologies either[71]. Accordingly, the traditional biopsy-first approach, followed by Sanger sequencing of clinically prioritised individual genes has been estimated to achieve genetic diagnosis in only approximately 11% of patients overall[53]. Further, incremental sequencing costs can exceed the costs of WES with targeted panel analysis[53] whilst the iterative process can prolong the diagnostic odyssey for individuals and reinforce diagnostic bias[14,37].
The impact and challenges of evolving NGS technologies
Targeted nuclear gene panels incrementally improve diagnosis compared to traditional single-gene Sanger sequencing, with rates reported between 6%-37% after mtDNA sequencing and dependent on the selected gene set and patient group, as summarised in Table 1[22,23,53,61,71-74]. However, the vast majority of patients remain undiagnosed. This approach focuses on commonly known disease genes and mutations, contributing less to the collective understanding of mitochondrial biology and disease. Unsurprisingly then, approaches utilising WES combined with mtDNA sequencing (either in advance, or incorporated into WES[75-77]) have further improved genetic diagnosis rates to between 35%-68%, depending on the selected patient group, as summarised in Table 2[15-21,23]. In one study, 31% of cases resolved through WES would have been missed using contemporaneous MitoCarta-based panels[15]. These results have included many novel disease genes and mutations, (43%-51% of cases in two paediatric studies[16,17]), thus expanding genotypic heterogeneity, whilst demonstrating greater phenotypic heterogeneity of known disease genes[3,23,78], highlighting the shortcomings of candidate gene approaches.
Diagnostic yield from NGS panel studies in mitochondrial disease
| Authors | Patients (n) | Age | Disease characteristics | Prior sequencing | Sequencing approach mtDNA (n; %) nDNA (n; %) | Nuclear genes | Yield: overall mtDNA % (n) [of solved] nDNA % (n) [of solved] | MD % (n) of solved cases | Novel % (n) of solved cases |
|---|---|---|---|---|---|---|---|---|---|
| Schoonen et al.[22] 2019 | 127 | Paed all Onset < 20 yo Most < 1 yo | All MD Bioc RCD | Nil | mtDNA, nDNA panel ± WES NGS mtDNA (M) (all) Panel (68%) WES (6%) | 136 genes (3 custom panels) | 6% (8/127); 75% (6/8) of WES 0% (0/127) 6% (8/127) [all solved] | 63% (5/8 pts) 62% (8/13 var) | 62% (8/13 var) |
| Plutino et al.[72] 2018 | 80 | Adult predom 70% adult 30% paed | All MD Clin/bioc/histology | NS | 2 step NGS NGS mtDNA screen (all) Panel: targeted (all) | 281 genes (custom) | 29% (23/80) 19% (15/80) [65% (15/23)] 10% (8/80) [35% (8/23)] | N/A (only analysed MD genes) | 40% (4/10 nDNA var) |
| Legati et al.[23] 2016 | 125 | Paed predom 62% < 1yo | All MD Clin/bioc | mtDNA analysis Single n genes | 2 step: panel ± WES N/A Panel (all); WES (8%) | 132 genes (custom) | 20% (25/125): 15% panel; 5% WES N/A 20% (25/125) 19/125 panel; 6/10 WES | 84% (21/25) | NS |
| Lieber et al.[73] 2013 | 102 | Mixed Range: 0-64 Mean: 27 yo | Def/HS 80% IS/LS 20% + control 18% | Varied mtDNA Single n genes | mtDNA + exome panel MitoExome mtDNA (all) MitoExome panel (all) | 1598 genes (MitoExome, expanded) | 23% (23/102); 17/18 control, 6/84 new 12% (12/102) [52% (12/23)] 11% (11/102) [48% (11/23)] | 50% (3/6 new diagnoses) | 17% (1/6 new diagnoses) |
| DaRe et al.[61] 2013 | 148 | Paed predom 83% < 18 yo Range 0-83 | Mixed Def/HS-MD 36% bioc RCD | Varied mtDNA Single n genes | Targeted exome panel N/A Panel: targeted (all) | 447 genes (custom) | 9% (13/148) N/A 9% (13/148) [all solved] | 31% (4/13 pts) | 48% (10/21 var) |
| Neveling et al.[53] 2013 | 44 | Mixed Mean 11.4 Range 2-30 | All MD Bioc (M/Fib) | mtDNA, POLG, ≤ 10 n genes | Targeted exome panel N/A Panel: targeted (all) | 211 genes (custom) | 16% (7/44) N/A 16% (7/44) [all solved] | 100% (7/7 pts) | NS |
| Calvo et al.[74] 2012 | 42 | Paed All NN/I | All MD Clin/Bioc RCD | NS | mtDNA + exome panel MitoExome mtDNA (all) MitoExome panel (all) | 1034 genes (MitoExome) | 31% (13/42) 2% (1/42) [8% (1/13)] 29% (12/42) [92% 12/13)] | 100% (13/13) | 23% (3/13 pts) |
| Vasta et al.[71] 2012 | 26 | Paed all Onset < 1 yo 88% | Mixed: Def, HS and LS-MD 2 + control | 10/26 mtDNA seq | Targeted NGS panel N/A NGS panel (all) | 908 genes | 27% (7/26); 2/2 control, 5/24 new N/A 27% (7/26) [all solved] | 71% (5/7 pts) | 62% (8/13 var) |
Diagnostic yield from WES studies in mitochondrial disease
| Authors | Patients n | Age | Disease characteristics | Prior sequencing | Sequencing approach mtDNA (n; %) nDNA (n; %) | Nuclear genes | Yield: overall mtDNA % (n) [of solved] nDNA % (n) [of solved] | MD % (n) of solved cases | Novel % (n) of solved cases |
|---|---|---|---|---|---|---|---|---|---|
| Theunissen et al.[15] 2018 | 117 | Paed predom Onset < 18 yo 77% | MD 74% (clin/bioc) NM 26% | Nil | 2 step NGS: NGS whole mtDNA (B ± U.M) (all) WES (94/117; 80%) | Unfiltered | 68% (80/117) 20% (23/117) [29% (23/80)] 49% (57/117) [71% (57/80)] | 73% (58/80) 23 mtDNA | 35 nDNA | NS |
| Puusepp et al.[20] 2018 | 28 | Paed all Onset < 7 yo NN/I predom | Def MD 14% HS-MD 57% I/LS-MD 29% | Targeted mtDNA Single n genes | WES + mtDNA Off-target (all) WES (all) | Unfiltered | 57% (16/28) 0% (0/28) [0% (0/16)] 57% (16/28) [100% (16/16)] | 25% (4/16) 75% Def MD 25% HS-MD 0% I/LS-MD | 70% (19/27 var) |
| Kohda et al.[21] 2016 | 142 | Paed all Onset < 15 yo 46% < 1 mo | All MD Bioc RCD | NS | mtDNA, WES, CGH LR-PCR mtDNA (all) WES (all) | Unfiltered | 35% (49/142) 7% (10/142) [20% (10/49)] 25% (35/142) [71% (35/49)] | 86% (42/49) | 61% (30/49 pts) 67% (40/60 var) |
| Pronicka et al.[17] 2016 | 113 | Paed all 42% NN | HS-MD 35% IS-MD 27% LS-MD 37% | Routine (NS) | WES (after routine) NS WES (all) | Unfiltered | 59% (67/113) (36% LS, 90% HS) 5% (6/113) [9% (6/67)] 54% (61/113) [91% (61/67)] | 70% (46/67 pts) 20% LS-MD 97% HS-MD | 51% (50/99 var) |
| Wortmann et al.[19] 2015 | 109 | Paed Young adult All < 27 yo | HS-MD 39% IS-MD 40% LS-MD 21% | mtDNA analysis CGH | 2 step: VP ± WES N/A WES (all) | VP (custom) 238 genes Unfiltered | 38% (42/109) (57% HS, 35% LS) N/A 38% (42/109) [all solved] | 62% (26/42) | NS |
| Ohtake et al.[16] 2014 | 104 | NS | All MD Bioc RCD | mtDNA analysis | WES (after mtDNA) N/A WES (all) | Unfiltered | 43% (45/104) N/A 43% (45/104) [all solved] | 100% (45/45) | 98% (44/45 pts) 17 variants, 27 genes |
| Taylor et al.[18] 2014 | 53 | Paed all 96% < 15 yo 66% < 1 yo | All MD Bioc MRCD | mtDNA analysis CGH | WES (after mtDNA) N/A WES (all) | Unfiltered | 60% (32/53) N/A 60% (32/53) [all solved] | 100% (32/32) | NS (12.5% novel genes) |
NGS technologies have dramatically accelerated the identification of novel mitochondrial disease genes and mutations, with around 15-20 new genes discovered annually over the past decade and more than 350 genes across the nuclear and mitochondrial genome implicated in disease[3,4,46]. Identification and functional validation of novel gene and mutation candidates have in turn provided novel insights into mitochondrial structure, function, dynamics, and mechanisms of disease[23,79]. Even early studies evaluating NGS technologies recognised their potential to revolutionise the diagnostic process for heterogeneous disorders, such as mitochondrial disease[42]. WES or WGS are already resolving many outstanding challenges associated with mitochondrial disease genetics, in turn improving patient care[21]. However, a distinction must be drawn between routine clinical genetic testing and the ongoing interchange between research and genetic diagnosis. The latter is critical for expanding the list of known pathogenic variants, improving the understanding of mitochondrial biology and disease, enabling refinement of the diagnostic pipeline and enhancing the routine interpretation of genetic variants. This is necessary to positively impact the evolution of genetic diagnosis from here, as well as inform clinical management, family planning and potential therapeutic avenues.
A further important benefit of comprehensive, non-targeted sequencing is the identification of pathogenic non-mitochondrial disease variants: mimics and phenocopies, especially neurological disorders and neuromuscular diseases, amongst other monogenic disorders, providing definitive genetic diagnosis, and, at times, important therapeutic options[56]. Depending on the cohort selection criteria for WES studies, proportions of (solved) cases attributable to mitochondrial diseases range from 25%-89%[15,17,19-21] underscoring the clinically important overlap with other monogenic disorders. Given the broad range of overlapping disorders to be considered, necessitating multiple sequential panels, the additional cost of exome sequencing is rapidly negated - and costs continue to decrease. In contrast to targeted gene panels, WES or WGS is applicable for most genetic disorders[80], can incorporate a virtual gene panel initially if desired, and expand if inconclusive, improving cost-effectiveness, and can be readily re-investigated[19].
Whilst the substantial benefits are evident, there are evolving challenges too. Vast amounts of data are generated by exome and genome sequencing. This presents important practical challenges for both sufficient, secure data storage and comprehensive and accurate analysis - particularly of the many variants of uncertain significance and novel variants requiring functional validation - as well as ethical questions pertaining to the identification and reporting of incidental findings. In 2015, the American College of Medical Genetics and Genomics and the Association for Molecular Pathology released revised guidelines informing sequence variant interpretation, incorporating limited guidelines for mitochondrial variant interpretation and noting specific associated challenges[81]. Despite this framework, interpretation remains challenging and inherently subjective, particularly for mtDNA variants[82]. Therefore, clinical and biochemical phenotyping remain important for successful utilisation and accurate interpretation of evolving sequencing techniques[19], with family trio sequencing incorporated where feasible, especially for paediatric cases and segregation studies in adults, to rapidly prioritise de novo variants. Increasing use of WES and WGS combined with evolving “omics” techniques, including metabolomics, proteomics and transcriptomics, are enabling further interrogation and evaluation of variants, generating data to inform variant prioritisation and assignment, pathophysiological insights and therapeutic options[83].
A proposed new approach
We suggest a genetics first diagnostic approach given the technical suitability of NGS for mitochondrial disease genetics and the expanding capability to reliably identify and call mitochondrial disease variants[26]. A genetics first diagnostic approach is also advocated for by others[56,57] and a proposed process is outlined in Figure 2 (adapted from Davis et al.[1]).
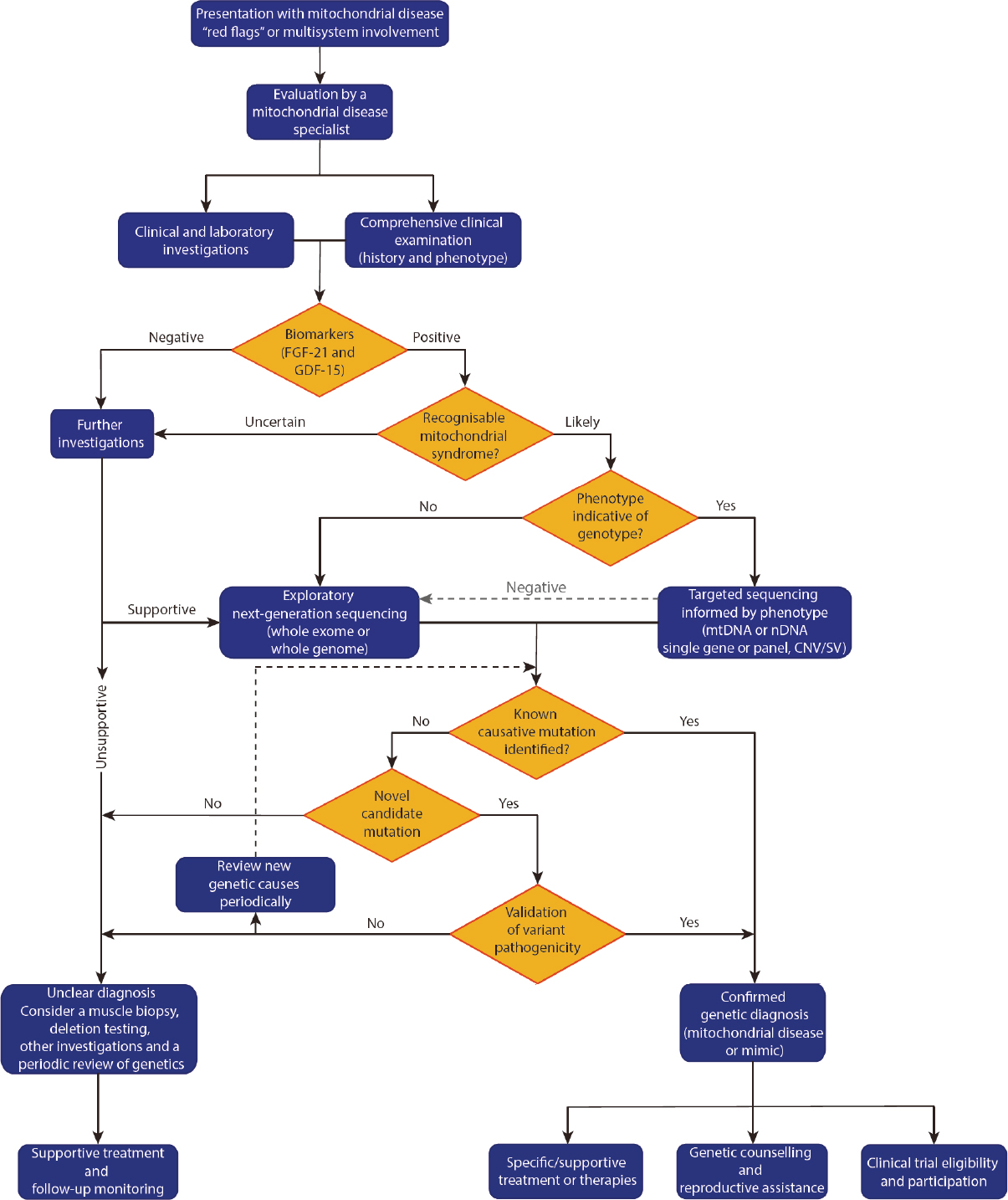
Figure 2. A proposed diagnostic approach for mitochondrial disease
The first stage aims to stratify the population for testing by answering two questions: (1) “is mitochondrial disease likely?” and, if so, (2) “is there a distinctive phenotype indicative of the genotype?” to inform the most appropriate genetic testing strategy. The next stage focuses on molecular diagnosis - either identifying a known pathogenic mutation, validating a novel mitochondrial-disease causing variant, or identifying a genetic phenocopy.
A careful and comprehensive history, including inheritance pattern where possible, together with comprehensive clinical examination, enables accurate clinical phenotyping and should be combined with tailored initial investigations to characterise organ involvement and form an initial clinical estimate of the likelihood of mitochondrial disease. Routine laboratory investigations, including those aimed at excluding infective or inflammatory processes and other mimics, should be undertaken alongside specific evaluation of serum lactate and pyruvate, creatine kinase (CK) and a urinary metabolic screen: indicators of disease but with limited sensitivity and specificity[54,66]. In adults, neuroimaging typically includes MRI of the brain (ideally with MR spectroscopy of CSF), and may demonstrate characteristic or non-specific patterns, or be normal[66,84-86]. Electroencephalogram, nerve conduction studies and electromyography may complement the initial clinical evaluation. Cardiac evaluation with electrocardiogram, 24-hour holter monitor and echocardiogram is critical to evaluate potentially life-threatening organ involvement and bedside ophthalmological examination may be augmented by retinal photography, or formal ophthalmological evaluation where appropriate.
The incorporation of biomarkers may aid clinical stratification (discussed below). If initial clinical evaluation and investigations are equivocal and/or biomarkers are negative, further supportive evidence for disease should be sought, prior to initiating comprehensive genetic testing. For example, the yield from detailed ophthalmological evaluation is high[87], with findings often specific for mitochondrial disease, whereas other investigations, such as GI motility, although predictive of a positive genetic diagnosis when present, are less specific for mitochondrial diseases or a particular genetic culprit[88].
The combination of suggestive clinical features, inheritance and initial investigations, together with positive biomarkers, should prompt the clinician to progress to genetic evaluation. Where a classical phenotype suggests a deletion syndrome, or one of a restricted group of causative genes or mutations, established targeted sequencing approaches in an appropriate tissue source (deletions often require uroepithelium or muscle) are readily available, rapid and cost-effective. If targeted sequencing returns negative, and in the many instances where a specific genetic cause or candidate is not able to be proposed, a comprehensive sequencing approach encompassing all potentially causative genes should be considered (discussed further below).
If, after comprehensive bigenomic sequencing, a genetic diagnosis still cannot be established, a review of the clinical presentation, consideration for further investigations - including muscle biopsy for biochemical and enzymatic studies, and genetics (in post-mitotic tissue) - and a periodic review of genetic data should be undertaken, as bioinformatics pipelines, variant analysis and the catalogue of known disease genes and pathogenic mutations are rapidly evolving.
With this proposed approach, muscle biopsy is not omitted entirely. Rather, it is selectively utilised to achieve specific end-points. Scenarios where early incorporation of muscle biopsy may be relevant include the genetic diagnosis of mtDNA deletion syndromes, where less invasive sources (blood leukocytes/uroepithelium) have been unrevealing, and for consideration where there is substantial uncertainty regarding the underlying aetiology that warrants further evaluation before proceeding further toward definitive diagnosis. However, it should be noted that in the latter context, WES has demonstrated utility in patients with a lower pre-sequencing likelihood of mitochondrial disease - the “possible” rather than “probable” group - as it can identify coding variants causing mitochondrial disease and monogenic disease mimics[15,19,20]. Therefore, careful consideration should be given to whether invasive investigation is justified at this stage. Later incorporation of muscle biopsy may be relevant for evaluation and functional validation of identified novel variants[4], in cases where definitive genetic diagnosis is not forthcoming, for investigation using more disease-relevant post-mitotic tissues, including to interrogate mtDNA deletions[89] and/or histological and biochemical evidence of mitochondrial disease in the absence of genetic diagnosis.
The role of serum biomarkers
The addition of sensitive and specific serum biomarkers to the initial evaluation may aid stratification of genetic testing. Traditional and commonly tested serum biomarkers of mitochondrial disease include lactate, pyruvate, their ratio, and CK. However, results may vary substantially, depending on factors including activity, diet and sample handling[90] and they lack sufficient sensitivity and specificity for clinical utility in mitochondrial disease[91]. Recently, more sensitive and specific serum biomarkers have been identified, although there remains scope for improvement.
Elevated levels of fibroblast growth factor-21 (FGF-21) have been demonstrated in people with muscle-manifesting mitochondrial diseases, compared to non-mitochondrial disease and healthy controls[91-93]. Further research indicates FGF-21 levels best correlate with defects of mitochondrial translation and may be normal in defects of respiratory chain complexes or their assembly factors[93]. More recent functional studies of mitochondrial myopathy in human and mouse models demonstrate the crucial role of FGF-21 in the integrated mitochondrial stress response (ISRmt), activating the systemic stress response and inducing systemic metabolic consequences[94]. However, FGF-21 levels can also be elevated in non-mitochondrial diseases, including some non-mitochondrial myopathies, cancer, obesity, renal disease, diabetes and liver disease[90], limiting diagnostic utility independent of clinical context.
The elevation of growth differentiation factor 15 (GDF-15) was identified in Thymidine Kinase (TK2)-related mitochondrial disease[95]. It was further evaluated in patient cohorts with mitochondrial and non-mitochondrial diseases[96-100], with some suggestion that GDF-15 levels may correlate with disease severity[97]. Davis and colleagues demonstrated improved diagnostic sensitivity and a higher diagnostic odds ratio for GDF-15 compared to FGF-21, noting that GDF-15 was potentially more broadly applicable than FGF-21[96]. This was followed by suggestion of better correlation with mitochondrial translation and mtDNA maintenance defects[90]. GDF-15 may also be elevated, albeit to a lesser degree, in non-mitochondrial muscle and metabolic diseases, pregnancy, diabetes, cancer, liver fibrosis and cardiovascular disease[90,101], and may reflect oxidative stress[101].
Both FGF-21 and GDF-15 are non-invasive serum assays, and although not independently diagnostic[101], offer superior utility to classical biomarkers[99]. They therefore complement clinical evaluation and can better inform decision making on subsequent costly tests such as NGS, whilst noting clinically relevant limitations.
Which sequencing approach?
Although targeted NGS panels achieved early advances in genetic diagnosis, there are clear benefits of WES or WGS approaches. Both generate vastly more data and demand upfront resources for analysis, although costs are rapidly decreasing, and can simultaneously analyse mtDNA, identify novel disease genes and variants, as well as monogenic phenocopies.
WES has demonstrated increased diagnostic yield in mitochondrial disease studies as outlined above[15-23], although it has frequently been utilised only for nuclear genome analysis following dedicated mtDNA genome sequencing. Off-target WES reads sufficiently capture mtDNA to assemble a mitochondrial genome[102] and analyse mtDNA variants with reasonable precision[75], owing to the abundance of mtDNA relative to nDNA. However, greater depth of coverage is required for reliable detection of low-heteroplasmy variants[76]. Dedicated mtDNA enrichment enables simultaneous analysis of mtDNA, with enhanced detection of low heteroplasmy variants, down to 8%[76]. Despite vast progress, however, a substantial proportion (30%-70%) of patients remain undiagnosed following WES[15-21]. Whilst this may reflect bioinformatic prioritisation or evolving analytic pipelines, there remain a number of insufficiencies in WES: coverage may be non-uniform and importantly limited in certain regions (especially G-C rich)[103] and indels and copy number variations may not be reliably identified[104]. Furthermore, PCR and mtDNA enrichment also introduce sequencing error and bias, the nature and extent of which depend on the selected kit and methods[103,105]. By definition, causative variants in non-coding regions are also omitted by WES. WGS can overcome all of these limitations to offer further utility, with promising early data in rare diseases[104] that may justify the modest additional cost.
PCR-free whole genome sequencing avoids sequencing error and biases introduced by library amplification, offering more consistent breadth and depth of coverage of coding regions[24] as well as covering the extensive non-coding regions. WGS can detect small and large chromosomal copy number variants[82], an increased proportion of single nucleotide variants and structural variants[24,25,104]. It also offers superior mtDNA coverage (1200-4000× with acceptable coverage depths of the nuclear genome, between 14-30×), allowing reliable detection of low-heteroplasmy variants, down to 2% or less[26,57]. Whilst analysis of mitochondrial variants presents unique challenges compared to interpretation of nuclear variants[106] which have more established bioinformatics pipelines, we have developed a novel dedicated tool, mityTM to offer automated, integrated mtDNA variant calling from WGS data[26]. Nuclear and mtDNA bioinformatics pipelines may be linked, facilitating simultaneous analysis of both nuclear and mitochondrial genomes from a single, minimally-invasive sample[26]. WGS therefore offers comprehensive, simultaneous bigenomic sequencing with superior mtDNA coverage depth and heteroplasmy sensitivity, whilst reducing introduced sequencing error and bias, and should therefore be the preferred sequencing option. Early WGS results from mitochondrial disease studies indicate the yield is at least equivalent for known variants, with potential for improved yield with novel variant identification and as analysis - especially of non-coding regions - evolves.
Conclusion: a minimally invasive, streamlined approach to mitochondrial disease genetic diagnosis
Despite significant advances in technology and understanding of mitochondrial biology over recent decades, the diagnosis of mitochondrial disease continues to present a challenge to the clinician and a large proportion of cases remain undiagnosed. Whilst the prevailing diagnostic paradigm advocates a “function-to-gene” approach centred on muscle biopsy, the substantial benefits of a “genetics-first” approach justify a paradigm shift. Such an approach, as proposed here, incorporating clinical evaluation, serum biomarker stratification and early bigenomic WGS, offers the potential to streamline a less invasive diagnostic process for patients, improve diagnostic yield, inform individual prognosis and the collective understanding of mitochondrial biology and ultimately pave the way for substantial therapeutic advances.
Declarations
Authors’ contributionsMade substantial contributions to data interpretation, conception and design of the work, revision of the manuscript: Watson E, Davis R, Sue CM
Drafting: Watson E
Made technical support: Davis R, Sue CM
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
2. Chinnery PF. Mitochondrial disorders overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle; 1993.
3. McCormick EM, Zolkipli-Cunningham Z, Falk MJ. Mitochondrial disease genetics update: recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr Opin Pediatr 2018;30:714-24.
4. Thompson K, Collier JJ, Glasgow RIC, Robertson FM, Pyle A, et al. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J Inherit Metab Dis 2020;43:36-50.
5. Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 2015;77:753-9.
6. Vandebona H, Mitchell P, Manwaring N, Griffiths K, Gopinath B, et al. Prevalence of mitochondrial 1555A-->G mutation in adults of European descent. N Engl J Med 2009;360:642-4.
7. Manwaring N, Wang JJ, Mitchell P, Sue CM. Mitochondrial DNA disease prevalence: still underrecognized? Ann Neurol 2008;64:471. author reply 471-2
8. Manwaring N, Jones MM, Wang JJ, Rochtchina E, Howard C, et al. Population prevalence of the MELAS A3243G mutation. Mitochondrion 2007;7:230-3.
11. Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, et al. Mitochondrial diseases. Nat Rev Dis Primers 2016;2:16080.
12. Munnich A, Rötig A, Chretien D, Cormier V, Bourgeron T, et al. Clinical presentation of mitochondrial disorders in childhood. J Inherit Metab Dis 1996;19:521-7.
13. Liang C, Ahmad K, Sue CM. The broadening spectrum of mitochondrial disease: shifts in the diagnostic paradigm. Biochim Biophys Acta 2014;1840:1360-7.
14. Grier J, Hirano M, Karaa A, Shepard E, Thompson JLP. Diagnostic odyssey of patients with mitochondrial disease: results of a survey. Neurol Genet 2018;4:e230.
15. Theunissen TEJ, Nguyen M, Kamps R, Hendrickx AT, Sallevelt S, et al. Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front Genet 2018;9:400.
16. Ohtake A, Murayama K, Mori M, Harashima H, Yamazaki T, et al. Diagnosis and molecular basis of mitochondrial respiratory chain disorders: exome sequencing for disease gene identification. Biochimi Biophys Acta 2014;1840:1355-9.
17. Pronicka E, Piekutowska-Abramczuk D, Ciara E, Trubicka J, Rokicki D, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med 2016;14:174.
18. Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014;312:68-77.
19. Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis 2015;38:437-43.
20. Puusepp S, Reinson K, Pajusalu S, Murumets Ü, Õiglane-Shlik E, et al. Effectiveness of whole exome sequencing in unsolved patients with a clinical suspicion of a mitochondrial disorder in Estonia. Mol Genet Metab Rep 2018;15:80-9.
21. Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet 2016;12:e1005679.
22. Schoonen M, Smuts I, Louw R, Elson JL, van Dyk E, et al. Panel-based nuclear and mitochondrial next-generation sequencing outcomes of an ethnically diverse pediatric patient cohort with mitochondrial disease. J Mol Diagn 2019;21:503-13.
23. Legati A, Reyes A, Nasca A, Invernizzi F, Lamantea E, et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim Biophys Acta 2016;1857:1326-35.
24. Meienberg J, Bruggmann R, Oexle K, Matyas G. Clinical sequencing: is WGS the better WES? Hum Genet 2016;135:359-62.
25. Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A 2015;112:5473-8.
26. Puttick C, Kumar KR, Davis RL, Pinese M, Thomas DM, et al. A highly sensitive mitochondrial variant analysis pipeline for whole genome sequencing data. bioRxiv 2019:852210.
27. Ernster L, Ikkos D, Luft R. Enzymic activities of human skeletal muscle mitochondria: a tool in clinical metabolic research. Nature 1959;184:1851-4.
28. Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest 1962;41:1776-804.
29. Engel WK, Cunningham GG. Rapid examination of muscle tissue. an improved trichrome method for fresh-frozen biopsy sections. Neurology 1963;13:919-23.
30. Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol 1984;16:481-8.
31. Fukuhara N, Tokiguchi S, Shirakawa K, Tsubaki T. Myoclonus epilepsy associated with ragged-red fibres (mitochondrial abnormalities): disease entity or a syndrome? Light-and electron-microscopic studies of two cases and review of literature. J Neurol Sci 1980;47:117-33.
32. Schon EA, Hirano M, DiMauro S. Mitochondrial encephalomyopathies: clinical and molecular analysis. J Bioenerg Biomembr 1994;26:291-9.
33. Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, et al. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002;59:1406-11.
34. Naviaux RK. Developing a systematic approach to the diagnosis and classification of mitochondrial disease. Mitochondrion 2004;4:351-61.
35. Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, et al. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord 1992;2:125-35.
36. DiMauro S, Garone C. Historical perspective on mitochondrial medicine. Dev Disabil Res Rev 2010;16:106-13.
37. Koenig MK. Presentation and diagnosis of mitochondrial disorders in children. Pediatr Neurol 2008;38:305-13.
38. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, et al. Sequence and organization of the human mitochondrial genome. Nature 1981;290:457-65.
39. Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988;242:1427-30.
40. Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988;331:717-9.
41. Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990;348:651-3.
42. McCormick E, Place E, Falk MJ. Molecular genetic testing for mitochondrial disease: from one generation to the next. Neurotherapeutics 2013;10:251-61.
43. Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 2016;44:D1251-7.
44. Frazier AE, Thorburn DR, Compton AG. Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J Biol Chem 2019;294:5386-95.
45. Craven L, Alston CL, Taylor RW, Turnbull DM. Recent advances in mitochondrial disease. Annu Rev Genomics Hum Genet 2017;18:257-75.
47. Zeviani M, Bresolin N, Gellera C, Bordoni A, Pannacci M, et al. Nucleus-driven multiple large-scale deletions of the human mitochondrial genome: a new autosomal dominant disease. Am J Hum Genet 1990;47:904-14.
48. Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet 1995;11:144-9.
49. Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve Suppl 1995;3:S107-12.
50. Chin J, Marotta R, Chiotis M, Allan EH, Collins SJ. Detection rates and phenotypic spectrum of m.3243A>G in the MT-TL1 gene: a molecular diagnostic laboratory perspective. Mitochondrion 2014;17:34-41.
51. Nesbitt V, Pitceathly RD, Turnbull DM, Taylor RW, Sweeney MG, et al. The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation--implications for diagnosis and management. J Neurol Neurosurg Psychiatry 2013;84:936-8.
52. Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol 2016;79:190-203.
53. Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg EJ, et al. A Post-Hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat 2013;34:1721-6.
54. Wortmann SB, Mayr JA, Nuoffer JM, Prokisch H, Sperl W. A guideline for the diagnosis of pediatric mitochondrial disease: the value of muscle and skin biopsies in the genetics era. Neuropediatrics 2017;48:309-14.
55. Kumar KR, Cowley MJ, Davis RL. Next-generation sequencing and emerging technologies. Semin Thromb Hemost 2019;45:661-73.
56. Niyazov DM, Kahler SG, Frye RE. Primary mitochondrial disease and secondary mitochondrial dysfunction: importance of distinction for diagnosis and treatment. Mol Syndromol 2016;7:122-37.
57. Raymond FL, Horvath R, Chinnery PF. First-line genomic diagnosis of mitochondrial disorders. Nat Rev Genet 2018;19:399-400.
58. Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med 2015;17:689-701.
59. Sue CM, Quigley A, Katsabanis S, Kapsa R, Crimmins DS, et al. Detection of MELAS A3243G point mutation in muscle, blood and hair follicles. J Neurol Sci 1998;161:36-9.
60. Broomfield A, Sweeney MG, Woodward CE, Fratter C, Morris AM, et al. Paediatric single mitochondrial DNA deletion disorders: an overlapping spectrum of disease. J Inherit Metab Dis 2015;38:445-57.
61. DaRe JT, Vasta V, Penn J, Tran NTB, Hahn SH. Targeted exome sequencing for mitochondrial disorders reveals high genetic heterogeneity. BMC Med Genet 2013;14:118.
62. Rodenburg RJ, Schoonderwoerd GC, Tiranti V, Taylor RW, Rötig A, et al. A multi-center comparison of diagnostic methods for the biochemical evaluation of suspected mitochondrial disorders. Mitochondrion 2013;13:36-43.
64. Gellerich FN, Mayr JA, Reuter S, Sperl W, Zierz S. The problem of interlab variation in methods for mitochondrial disease diagnosis: enzymatic measurement of respiratory chain complexes. Mitochondrion 2004;4:427-39.
65. Medja F, Allouche S, Frachon P, Jardel C, Malgat M, et al. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009;9:331-9.
66. Dimmock DP, Lawlor MW. Presentation and diagnostic evaluation of mitochondrial disease. Pediatr Clin North Am 2017;64:161-71.
68. Suomalainen A. Biomarkers for mitochondrial respiratory chain disorders. J Inherit Metab Dis 2011;34:277-82.
69. Oglesbee D, Freedenberg D, Kramer KA, Anderson BD, Hahn SH. Normal muscle respiratory chain enzymes can complicate mitochondrial disease diagnosis. Pediatr Neurol 2006;35:289-92.
70. Stenton SL, Prokisch H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays biochem 2018;62:399-408.
71. Vasta V, Merritt JL 2nd, Saneto RP, Hahn SH. Next-generation sequencing for mitochondrial diseases: a wide diagnostic spectrum. Pediatr Int 2012;54:585-601.
72. Plutino M, Chaussenot A, Rouzier C, Ait-El-Mkadem S, Fragaki K, et al. Targeted next generation sequencing with an extended gene panel does not impact variant detection in mitochondrial diseases. BMC Med Genet 2018;19:57.
73. Lieber DS, Calvo SE, Shanahan K, Slate NG, Liu S, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology 2013;80:1762-70.
74. Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 2012;4:118ra10.
75. Wagner M, Berutti R, Lorenz-Depiereux B, Graf E, Eckstein G, et al. Mitochondrial DNA mutation analysis from exome sequencing - A more holistic approach in diagnostics of suspected mitochondrial disease. J Inherit Metab Dis 2019;42:909-17.
76. Falk MJ, Pierce EA, Consugar M, Xie MH, Guadalupe M, et al. Mitochondrial disease genetic diagnostics: optimized whole-exome analysis for all MitoCarta nuclear genes and the mitochondrial genome. Discov Med 2012;14:389-99.
77. Abicht A, Scharf F, Kleinle S, Schön U, Holinski-Feder E, et al. Mitochondrial and nuclear disease panel (Mito-aND-Panel): combined sequencing of mitochondrial and nuclear DNA by a cost-effective and sensitive NGS-based method. Mol Genet Genomic Med 2018;6:1188-98.
78. Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol 2017;241:236-50.
79. Murayama K, Shimura M, Liu Z, Okazaki Y, Ohtake A. Recent topics: the diagnosis, molecular genesis, and treatment of mitochondrial diseases. J Hum Genet 2019;64:113-25.
80. Stark Z, Tan TY, Chong B, Brett GR, Yap P, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 2016;18:1090-6.
81. Richards S, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.
82. McCormick EM, Muraresku CC, Falk MJ. Mitochondrial genomics: a complex field now coming of age. Curr Genet Med Rep 2018;6:52-61.
83. Khan S, Ince-Dunn G, Suomalainen A, Elo LL. Integrative omics approaches provide biological and clinical insights: examples from mitochondrial diseases. J Clin Invest 2020;130:20-8.
84. Mascalchi M, Montomoli M, Guerrini R. Neuroimaging in mitochondrial disorders. Essays Biochem 2018;62:409-21.
85. Gropman AL. Neuroimaging in mitochondrial disorders. Neurotherapeutics: the journal of the American Society for Experimental. NeuroTherapeutics 2013;10:273-85.
86. Saneto RP, Friedman SD, Shaw DWW. Neuroimaging of mitochondrial disease. Mitochondrion 2008;8:396-413.
87. Ahmad K, Tan K, Sue C. The neuro-ophthalmology of mitochondrial disease with a particular focus on the morphology of the optic nerve head. J Clin Neurosci 2014;21:2043.
88. Finsterer J, Frank M. Gastrointestinal manifestations of mitochondrial disorders: a systematic review. Therap Adv Gastroenterol 2017;10:142-54.
89. Rahman S, Poulton J, Marchington D, Suomalainen A. Decrease of 3243 A-->G mtDNA mutation from blood in MELAS syndrome: a longitudinal study. Am J Hum Genet 2001;68:238-40.
90. Boenzi S, Diodato D. Biomarkers for mitochondrial energy metabolism diseases. Essays Biochem 2018;62:443-54.
91. Davis RL, Liang C, Edema-Hildebrand F, Riley C, Needham M, et al. Fibroblast growth factor 21 is a sensitive biomarker of mitochondrial disease. Neurology 2013;81:1819-26.
92. Suomalainen A. Fibroblast growth factor 21: a novel biomarker for human muscle-manifesting mitochondrial disorders. Expert Opin Med Diagn 2013;7:313-7.
93. Lehtonen JM, Forsström S, Bottani E, Viscomi C, Baris OR, et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 2016;87:2290-9.
94. Forsström S, Jackson CB, Carroll CJ, Kuronen M, Pirinen E, et al. Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab 2019;30:1040-54.e7.
95. Kalko SG, Paco S, Jou C, Rodríguez MA, Meznaric M, et al. Transcriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genomics 2014;15:91.
96. Davis RL, Liang C, Sue CM. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology 2016;86:2010-5.
97. Yatsuga S, Fujita Y, Ishii A, Fukumoto Y, Arahata H, et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann Neurol 2015;78:814-23.
98. Ji X, Zhao L, Ji K, Zhao Y, Li W, et al. Growth differentiation factor 15 Is a novel diagnostic biomarker of mitochondrial diseases. Mol Neurobiol 2017;54:8110-6.
99. Montero R, Yubero D, Villarroya J, Henares D, Jou C, et al. GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS One 2016;11:e0148709.
100. Poulsen NS, Madsen KL, Hornsyld TM, Eisum AV, Fornander F, et al. Growth and differentiation factor 15 as a biomarker for mitochondrial myopathy. Mitochondrion 2020;50:35-41.
101. Tsygankova PG, Itkis YS, Krylova TD, Kurkina MV, Bychkov IO, et al. Plasma FGF-21 and GDF-15 are elevated in different inherited metabolic diseases and are not diagnostic for mitochondrial disorders. J Inherit Metab Dis 2019;42:918-33.
102. Picardi E, Pesole G. Mitochondrial genomes gleaned from human whole-exome sequencing. Nat Methods 2012;9:523-4.
103. Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, et al. Characterizing and measuring bias in sequence data. Genome Biol 2013;14:R51.
104. Posey JE. Genome sequencing and implications for rare disorders. Orphanet J Rare Dis 2019;14:153.
105. Barbitoff YA, Polev DE, Glotov AS, Serebryakova EA, Shcherbakova IV, et al. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci Rep 2020;10:2057.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Watson E, Davis R, Sue CM. New diagnostic pathways for mitochondrial disease. J Transl Genet Genom 2020;4:188-202. http://dx.doi.org/10.20517/jtgg.2020.31
AMA Style
Watson E, Davis R, Sue CM. New diagnostic pathways for mitochondrial disease. Journal of Translational Genetics and Genomics. 2020; 4(3): 188-202. http://dx.doi.org/10.20517/jtgg.2020.31
Chicago/Turabian Style
Watson, Eloise, Ryan Davis, Carolyn M. Sue. 2020. "New diagnostic pathways for mitochondrial disease" Journal of Translational Genetics and Genomics. 4, no.3: 188-202. http://dx.doi.org/10.20517/jtgg.2020.31
ACS Style
Watson, E.; Davis R.; Sue CM. New diagnostic pathways for mitochondrial disease. J. Transl. Genet. Genom. 2020, 4, 188-202. http://dx.doi.org/10.20517/jtgg.2020.31
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 63 clicks
Cite This Article 63 clicks

 Like This Article 31
likes
Like This Article 31
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.