
Human induced pluripotent cells in personalized treatment of monogenic epilepsies
 0
0Abstract
The broad application of next-generation sequencing in genetic diagnostics opens up vast possibilities for personalized treatment of patients with genetic disorders including monogenic epilepsies. To translate genetic findings into personalized medicine, mechanistic studies of the individual pathogenic variants and drug screening in patient-specific in vitro models are very crucial. Recently, human induced pluripotent stem cell (hiPSC) technologies have made it possible to generate patient-specific pluripotent cells, which can be directed to differentiate into any given cell type. These hiPSCs are ideal for generating neurons to investigate specific neurological/neurodevelopmental disorders. While two-dimensional single-cell models of hiPSC-derived neurons provide reliable investigation of synaptic transmission and plasticity, cerebral organoids are superior in regard to functional characterization and the study of cell-cell interactions in three-dimensional structures. In this review, we focus on monogenic epilepsies and discuss the application of hiPSC models in personalized drug treatment based on the patient’s specific genetic variants.
Keywords
Introduction
Epilepsy is one of the most common neurological disorders[1], with a prevalence of 4-8 per 1000 and a lifetime risk of 3%[2]. Epilepsy is not a single disease entity, but rather a generic name used for diseases and syndromes, where abnormal brain activity causes seizures[3,4]. The underlying pathomechanisms of epilepsies are largely unknown[5]; however, genetic factors play a major role, either as variants in single genes causing monogenic epilepsies or as polygenic risk factors in common epilepsies. Like all other diseases, epilepsy has benefited from the genomic sequencing revolution in medicine. Next-generation sequencing-based methods such as targeted gene panels and exome sequencing are now routine analyses in many countries. It is estimated that these tests can provide a genetic diagnosis in up to 30% of patients with early-onset epilepsies[6-9]. Parallel to the increasing number of patients with genetically verified diagnosis, the demand for more accurate and targeted drug treatment also increases. The increasing number of patients with a precise genetic diagnosis enables researchers and clinicians to compare cohorts of patients with the same genetic defect and evaluate their treatment response to antiepileptic drugs (AEDs). These efforts along with functional studies have led to more precise treatment strategies in a subset of monogenic epilepsies, mainly ion channel disorders (channelopathies)[7,8,10]. The first-line treatment for most epilepsies is medication, and the choice of AED is primarily based on the epilepsy syndrome/seizure type and the age of the patient, while the underlying etiology plays only a minor role. However, more than 30 AEDs are available, and a therapy targeting the functional consequence of a given genetic variant without going through the odyssey of trial-error drug applications is crucial. In channelopathies, the drug of choice depends on whether a given genetic defect is a gain-of-function (GoF) or loss-of-function (LoF) variant, (activating vs. inactivating variant) leading to an imbalance in the function of inhibitory (e.g., GABAergic) or excitatory (e.g., glutamatergic) neurons. Patient-specific in vitro models, such as neurons differentiated from human-induced pluripotent stem cells (hiPSCs) with a given pathogenic variant can provide a tool for understanding the function of the gene variant and serve as drug screening platforms for targeted therapy of the patients. In this study, we focus on the application of iPSC-based functional studies and drug screening possibilities in monogenic epilepsies.
Epileptic channelopathies
Ion channels can broadly be classified as either voltage-gated or ligand-gated, depending on whether the stimulus for their activation is a change in the membrane potential or a chemical messenger such as a neurotransmitter, respectively. The role of ion channels in neuronal excitability is well established, and channelopathies include diseases of the central nervous system (CNS), and cardiovascular, respiratory, urinary, endocrine and immune systems. CNS-related channelopathies include familial hemiplegic migraine, episodic ataxia and various types of epilepsies[11].
Channelopathies account for a substantial fraction of epilepsy syndromes ranging from severe infantile developmental and epileptic encephalopathies to relatively benign focal epilepsies. More than 20 ion channel genes have so far been associated with epilepsy, and the majority of the most common epilepsy genes encode either voltage-gated sodium (SCN1A, SCN2A, SCN8A), potassium (KCNQ2, KCNQ3, KCNT1, KCNB1, KCNA2) or calcium channels (CACNA1A)[12]. Recent molecular genetic advances have contributed to our understanding of the pathophysiological mechanisms underlying these epilepsies. Careful clinical assessment and appropriate genetic tests and functional studies enable an accurate diagnosis, which is consequential for genetic counseling and guiding treatment options. Recently, some evidence emerged that dysfunctional channels can be specifically targeted by drugs acting on them[7,8,13]. However, different variants in the same gene can lead to either LoF or GoF, and it is challenging to predict the functional outcome of a given variant, which hampers choosing the right AED[7,8,14-16]. The combination of phenotypic, genetic and functional studies has resulted in remarkable advances in understanding the molecular and cellular disease mechanisms, and to find a steadily increasing number of tailored treatments. This translational road, from bed to bench and back to the patients, can be often realized by repurposed drugs (i.e., those that are already licensed for a different indication), for specific disease-related targets or pathways, thereby providing an immediate benefit for affected patients[7,8,13]. However, knowing the functional effect (LoF or GoF) of a variant is crucial and can assist in avoiding ineffective or even disease-aggravating treatments. For instance, individuals with GoF variants in SCN2A/3A/8A respond well to treatment with sodium channel blockers such as phenytoin, carbamazepine and oxcarbazepine, whereas the clinical picture of the individuals with LoF variants in SCN1A/2A/8A worsens with the same drugs[7,8].
To apply stratified treatment options for individual patients, it is of great advantage to determine the functional effect of the variant and potentially test drug efficacy in patient-specific systems.
Pathophysiology of epilepsy
The pathophysiological mechanisms underlying epileptiform activity and seizures are hyperexcitability of cortical neurons and hypersynchronous simultaneous firing of numerous neurons, which is considered to be caused by an imbalance between inhibitory (GABAergic) and excitatory (glutamatergic) neurotransmission. Hyperexcitability of individual neurons can result from different mechanisms including decreased inhibitory or increased excitatory neurotransmission, alterations in voltage-gated ion channels, or alterations of intra- or extracellular ion concentrations favoring membrane depolarization and leading to epileptiform activity and seizures[17]. However, epileptogenesis is far more complex, and generation of epileptiform activity and seizures may include many other processes such as cellular diversity, synaptic spatiotemporal dynamics of interneuronal connectivity, synaptic reorganization of neuronal microcircuits, modifying network synchrony and brain oscillations[18]. Any brain region can potentially generate a seizure if the net excitation in a cortical area exceeds the net inhibition, and each step in seizure initiation, propagation, and termination is ultimately governed by the balance between excitation and inhibition[17]. Most currently available AEDs are aimed at restoring the excitatory/inhibitory balance either by decreasing excitation or by increasing inhibition[19].
Experimental models of epilepsy
Recent studies have identified > 500 epilepsy-associated genes, among which ion channel genes are predominant[6,20,21]. To investigate disease pathology and assess optimal treatment options for monogenic epilepsies, a wide range of cellular and animal models including fly, worm, zebrafish and rodent models have been developed[22]. In this review, we mainly focus on the cellular models [non-neuronal and neuronal models, and human stem cell-derived two-dimensional (2D) and three-dimensional (3D) models], which have clearly increased our knowledge on the functional consequences of genetic variants in, for example, ion channel genes, the pathophysiology of epilepsy, and predicting drug efficacies. However, the knowledge gained has also raised many new questions to be answered. One of the crucial questions is how both GoF and LoF variants of the same channel gene can cause epilepsy, as exemplified by variants detected in SCN2A, SCN8A, KCNQ2, KCNQ3, KCNA2, CACNA1A and GRIN2A.
Cellular expression systems utilizing human embryonic kidney cells (HEK293), Xenopus laevis oocytes or Chinese hamster cells, are widely used as high throughput platforms for determining the biophysical consequences of genetic variants compared to the wild-type. The observed functional effects of the variants on ion channels often include shifts in activation and inactivation curves, changes in overall current amplitudes and altered persistent currents and kinetics of inactivation[23]. Currently, the gold standard for investigating the functional consequence of a certain variant is to express the mutant channel in Xenopus oocytes and use two-electrode voltage clamp methods to assess the function[24]. Voltage clamping allows the monitoring of minute changes in the electric currents across membranes. It is a fast and low-cost screening system where the biophysical consequences can be studied using heterologous expression systems that are not contaminated by endogenous channels[22]. Furthermore, Xenopus oocytes have the advantages of small size, durability and effectiveness in exogenous protein expression. These expression systems have not only been used to study the electrophysiological consequences of genetic variants but also to investigate their pharmacological characteristics[13,25]. Xenopus oocyte models have, for instance, been used to study the effect of the potassium channel blocker quinidine on GoF of KCNT1 channels[13], and neuroblastoma ND7/23 cells have been used to study the effect of the sodium channel blocker phenytoin on GoF of SCN8A channels[25]. Both studies have successfully shown that the investigated drugs could reverse the variant specific GoF, which indicates that these conditions may respond to the targeted therapies. Although these cellular expression systems are extremely valuable, a clear drawback is that they only allow theoretical predictions as to how neuronal behavior might be changed by the variants[26]. It is therefore crucial to test the dysfunctional proteins in a human neuronal specific context, which can mimic human physiological conditions to a better extent.
hiPSC models in epilepsy disorders
Patient hiPSCs have successfully been used to model several different monogenic epilepsies including epilepsy in infancy with migrating focal seizures[27], tuberous sclerosis[28], PCDH19 girls clustering epilepsy[29], Dravet syndrome[30,31] and Angelman syndrome[32,33]. On the one hand, these studies have provided novel mechanistic insights and served as proof of principle platforms for using hiPSCs to study epilepsy, but on the other hand, they have also highlighted the potential limitations. In the following, we will discuss the hiPSC models of Dravet syndrome as an example.
Dravet syndrome
Dravet syndrome is a severe developmental and epileptic encephalopathy (DEE) caused by LoF variants in SCN1A, encoding the voltage-gated sodium channel Nav1.1. The syndrome is characterized by prolonged fever-induced hemi-clonic or generalized tonic-clonic seizures (GTCS) with onset at around 6 months of age[7,34]. Over time, the patients also develop afebrile seizures including GTCS, atypical absences, and focal and myoclonic seizures. Development is often normal at onset; however, during the course of the disease, most patients develop moderate-to-severe intellectual disability, behavioral problems, sleep disturbances, and both gross and fine motor impairment[7,34]. To understand the physiopathology of Dravet syndrome, hiPSC models were generated[30,31]. In one of these studies, patient-derived neurons could not respond adequately to high-intensity stimulation, which suggested an impairment in the function of GABAergic neurons and would explain the epileptogenesis in the syndrome[30]. In another study neurons derived from patient hiPSCs displayed increased sodium currents in both bipolar- and pyramidal-shaped neurons, accompanied by spontaneous bursting and other evidence of hyperexcitability[31]. In addition, in hiPSC-derived neurons, the expression of tyrosine hydroxylase mRNA and subsequent protein abundance was increased, which might indicate that pathogenic variants in SCN1A could change the dopamine system and responses[35].
Measuring electrophysiological properties of neurons and cellular networks
The electrophysiological properties of differentiated neurons can be measured to clarify the impact of the genetic variant on the function of the protein/channel and the properties of the neural network in a disease and species-specific context using different systems. Furthermore, these systems can be used in drug screening to assess which drug(s) can reverse the electrophysiological imbalance of a differentiated neuron with a given pathogenic variant.
Voltage clamping allows monitoring of minute changes in the electric currents across membranes. 2D cellular models of excitatory/glutamatergic and inhibitory/GABAergic neurons are ideal for functional readouts. Two techniques commonly used to assess the electrophysiological properties of model systems are (1) single-cell patch-clamp electrophysiological recordings; and (2) multielectrode array (MEA) which is described below. The main difference between these techniques is that the former method measures synaptic activity of single neurons, while MEA measures electrophysological activity of neuronal networks. MEA uses several microelectrodes embedded in the surface of neuron cultures and allows the targeting of several sites in parallel to record and measure spontaneous firing of action potentials of neuronal networks. MEA has effectively been used for different approaches in epilepsy studies, such as predicting seizure liability at the early stage of preclinical studies, drug screening and drug safety[36,37]. Recently, utilizing MEA in combination with in vitro hiPSC models has effectively been applied to drug screening studies such as frequency analysis, revealing prediction of toxicity and side effects of anticonvulsants and their mechanism of action. In addition, it has been demonstrated that hiPSC-derived astrocytes and neuron co-cultures mimic in vivo conditions better than monocultures of neurons. These observations suggest that such experimental setup may be considered suitable for toxicity prediction[38].
MEAs are generally easy to handle in terms of cell types in the cultures and reproducibility. Although, patient-derived neurons are investigated, the MEA system cannot achieve the complexity needed to understand the pathophysiology and cell type-specific communication between different types of neurons, astrocytes and oligodendrocytes in the brain. In contrast, MEA analyses of neurons could potentially be useful for large scale drug screening to investigate if certain drugs/compounds could restore abnormal network activity[37] in neurons. The use of MEAs in drug screening in epilepsy is still in its infancy and more studies are needed before MEAs can be used as a high-throughput screening platform for personalized medicine.
IPSC-derived neuronal cultures
Cell types used in hiPSC programming
Epilepsy models based on hiPSC technology have the potential to be a revolutionary platform for functional studies, and as drug screening and discovery tools. Figure 1 illustrates the overview of the potential of hiPSC in relation to epilepsy. The most common cell type in generating hiPSCs is skin fibroblasts cultured from skin biopsies. This procedure was developed in the mouse, where skin fibroblasts were successfully reprogrammed using the murine transcription factors associated with pluripotency of cells in the inner cell mass of mouse blastocysts. Via a trial-and-error approach four core transcription factors (Klf4, c-Myc, Oct4 and Sox2), which have the potency to convert adult mouse skin fibroblasts into mouse iPSCs were identified[39]. These findings were subsequently applied to human skin fibroblasts using the equivalent human transcription factors[40]. Later, it became possible to generate hiPSCs from other somatic cells including peripheral blood mononuclear cells (PBMCs), urinary epithelial cells and epithelial stem cells from hair follicles[41,42] Even though several protocols are established to generate hiPSCs from alternative tissues, skin fibroblasts are the most preferred cell type, mainly due to the well-established and optimized protocols[43]. However, this trend is changing, especially in case of young patients, for whom the most favored cell type are PBMCs, where their collection is far less invasive[44]. The pros and cons of the different cell types for hiPSC generation are summarized in Table 1[45-59]. These hiPSCs obtained via the different reprogramming protocols are the starting material to generate any given cell type in a patient-specific manner by targeted differentiation, including neurons affected by epileptic disorders.
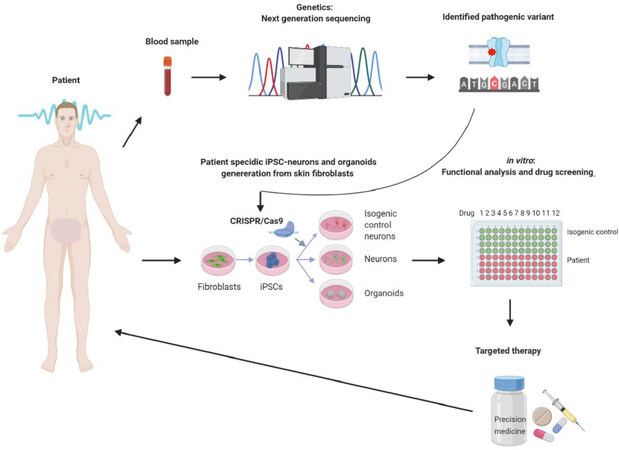
Figure 1. Schematic overview of the potential and utility of induced pluripotent stem cell (iPSC) in precision medicine. In the first step the likely pathogenic variant is identified using, e.g., next-generation sequencing technologies with DNA isolated from peripheral blood. In case of a variant without a known function loss-of-function or gain-of-function and/or for drug screening, primary fibroblast cultures are established from patient skin biopsies and reprogrammed into human induced pluripotent stem cell (hiPSC). The pathogenic variant can then be corrected using CRISPR/Cas9 gene editing, which generates isogenic controls for the patient hiPSCs with the pathogenic variant. Both hiPSCs are subsequently differentiated into relevant neurons (gluatamatergic or GABAergic in case of monogenic epilepsies) or cerebral organoids, which can be used in electrophysiological studies to determine the functional consequences and/or in drug screening
Comparison of different somatic cells as starting cell types
| Source | Initial cell type | Programming time | Reprogramming efficiency | Invasiveness | Availability | Ref. |
|---|---|---|---|---|---|---|
| Cord blood | Endothelial cells | 2-4 weeks | 2 | Yes | 1 | [46-48] |
| Peripheral blood | Mature T and myeloid cells
Peripheral blood mononuclear cells: lymphocytes, monocytes | 3-5 weeks | 1 | Yes | 2 | [49-51] |
| Hair | Keratinocytes | 2-3 weeks | 3 | No | 3 | [52-54] |
| Urine | Renal tubular/epithelial cells | 3 weeks | 3 | No | 3 | [55-57] |
| Skin | Fibroblasts | 3-5 weeks | 2 | Yes | 2 | [58-60] |
Alternatives to patient fibroblasts
Acquisition of patient tissues/cells is potentially challenging due to different reasons including invasiveness of the procedures (e.g., skin biopsies) and ethical regulations. An alternative to overcome this challenge is to use hiPSC lines from commercial sources. These hiPSC lines are from apparently normal individuals and are thoroughly characterized usually with whole-genome sequencing. With the recent development of CRISPR/Cas9 gene editing[60,61], it became possible to introduce any given gene variant into the genome of hiPSC. Furthermore, the unedited parent hiPSCs would function as an isogenic control with the same genomic background, while using patient cell lines necessitate age- and gender-matched controls, which are suboptimal due to the different genetic backgrounds, even when parents or siblings are used as controls. CRISPR/Cas9 gene editing has been successfully applied to hiPSCs, and it is especially useful in monogenic disorders. It also allows parallel introduction of different variants into the same parent hiPSCs, which gives an excellent opportunity to compare the functional consequences of the individual variants on the same genetic background without interference from specific genetic background noise of individuals.
Differentiation strategies-glutamatergic and GABAergic neurons
Since the main neuron types affected in epilepsy are GABAergic (inhibitory) and glutamatergic (excitatory) neurons. The main aim of a differentiation strategy is to achieve homogenous neuronal populations since heterogeneous neuronal populations will not be a true model of the disorder. Widely used differentiation strategies include (1) generation of embryoid bodies and isolation of neural rosette cells (neural tube-like structures); (2) use of small-molecule inhibitors; and (3) forced viral overexpression of transcription factors[62-64]. Even though the published neural cell differentiation protocols are effective and can be used to study specific neuronal subtypes, they are time-consuming and are still under development to further optimize the yield of desired subpopulations. Differentiation methodologies such as small-molecule inhibitors and neural rosette generation promote neuroectoderm development while repressing the other cell fate choices of the mesoderm and endoderm. This is achieved by a process referred to as dual SMAD inhibition. Proteins of the SMAD family are important signaling molecules in the TGFB (transforming growth factor beta) signaling pathway. The TGFB superfamily, including bone morphogenetic proteins (BMPs), nodal growth differentiation factor and activins, plays important roles in hiPSC maintenance and lineage commitment[65]. Specific inhibition of defined parts of TGFB signaling aids hiPSC differentiation into the neural and glial lineages. Two commonly used TGFB signaling inhibitors are Noggin and SB431542, which are referred to as dual SMAD inhibitors. Noggin inhibits BMP signaling and thereby abolishes trophectoderm and later ectoderm (skin) development, pushing the hiPSCs to differentiate into neuroectoderm and commonly referred to as neural progenitors cells (NPC). Noggin is applied in combination with SB431542, which inhibits activin and nodal signaling and suppresses mesoderm development[64]. This combined treatment with dual SMAD inhibitors became the gold standard for neural induction. Starting from this initial induction towards the neural fate, a battery of different protocols are available for different types of neurons.
For both the glutamatergic and GABAergic neurons, the initial step is neural induction from hiPSC to NPCs facilitated by dual SMAD inhibition. Subsequently, different protocols are applied to achieve glutamatergic [Figure 2] and GABAergic neurons.
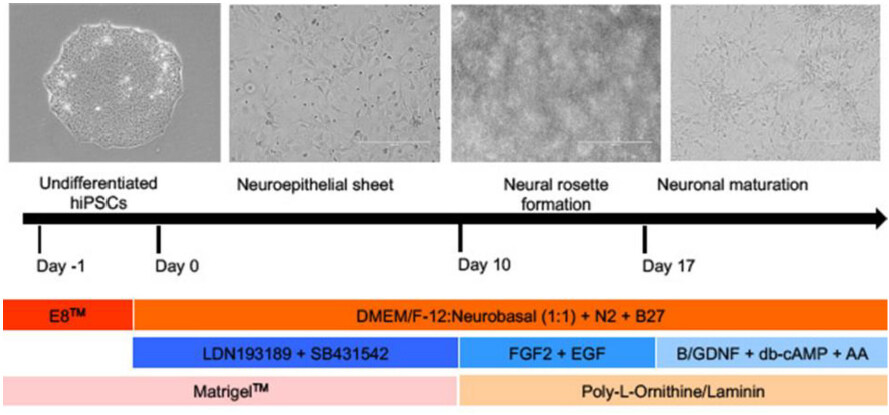
Figure 2. Brief protocol for directed differentiation of glutamatergic neurons from human induced pluripotent stem cell (hiPSC). Undifferentiated hiPSC are grown in E8 media (dark orange bar) to 80% confluency, whereupon they are exposed to the dual SMAD inhibitors (LDN193189 and SB431542) until day 10. Afterwards, proliferation of these neural progenitors (NPCs) are stimulated using fibroblast growth factor 2 (FGF2) and epidermal growth factor (EGF). A critical point is not to exceed passage 4 to avoid a shift to glial progenitors. After day 17 NPCs are matured in a medium containing brain-derived neurotrophic factor, glial cell-derived neurotrophic factor (GDNF), dibutyryl-cAMP (activates cAMP-dependent protein kinases) and ascorbic acid (AA) for a minimum of 5 weeks (blue horizontal bars). During this differentiation process, specific protein surface treatments are used in different stages to ensure cell expansion and retainment of stem cell identity or differentiation (pink and light orange bars). The basic growth and differentiation media are a 1:1 mixture of DMEM/F12 and Neuralbasal media, including N2 and B27 (orange horizontal bar), and extra factors are added at different time points (modified from[78,79])
A simplified differentiation procedure for excitatory glutamatergic or inhibitory GABAergic neurons is illustrated in Figure 3. Such monocultures are ideal for testing cell type-specific effects, pathologies and drug screening. However, it should be kept in mind that this model has limitations due to the fact that cells do not exist alone in their natural environment but in a complex network of different types of neurons. On the contrary, a clear advantage is that such neurons are more stressed in monocultures and display disease phenotypes more readily, which might be absent or take longer time to develop in co-culture systems. Nevertheless, complex systems such as cerebral organoids might be an advantage to study, for example, disease development in a more natural model system.
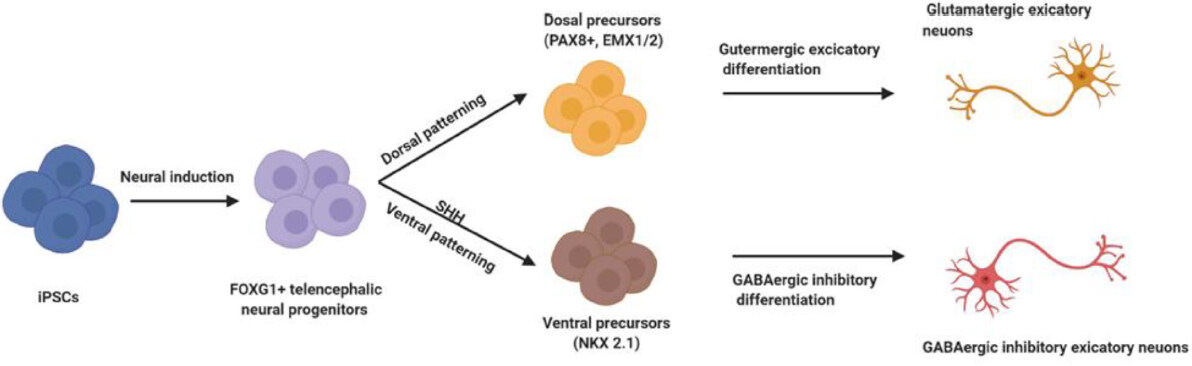
Figure 3. Schematic representation of differentiation procedures for generation of GABAergic and glutamatergic human induced pluripotent stem cell (hiPSC) neurons. Pluripotent hiPSCs can be induced via dual SMAD inhibition to produce telencephalic neural progenitors cells (NPCs) which are positive for FOXG1 - a brain-specific transcriptional repressor essential for the early development of the telencephalon - marking this population. Notably, FOXG1 gene is associated with the FOXG1 syndrome, which has previously been described as a congenital variant of Rett syndrome. In the further differentiation steps, the absence of a SHH (sonic hedgehog) agonist facilitates dorsal patterning of the NPCs, characterized by expression of the markers PAX8 (a human neuroectoderm cell fate determinant) and EMX1/2 (transcription factors expressed primarily in dorsal telencephalon. On the contrary, activation of SHH signaling via the addition of SSH agonists induces ventralization of NPCs, characterized by expression of NKX2-1 (a transcription factor that specifies ventral lineages during development). Dorsal progenitors can be further differentiated into glutamatergic neurons, whilst ventral precursors can be differentiated into GABAergic neurons
Limitations of hiPSC-based cell models
All these examples clearly illustrate the usefulness of patient-specific hiPSCs and their CRISPR/Cas9 gene-edited isogenic controls to model monogenic epilepsies. These are powerful models, especially if they are used in combination with patient data and animal models, and they will expand our knowledge of underlying cellular deficiencies observed in epilepsy. Nevertheless, several challenges remain and need to be addressed to further improve these hiPSC models. In terms of 2D monocultures, which are very applicable for large drug screens, the biggest challenge is heterogeneity of the generated neuronal populations. These heterogeneous populations are problematic if the disease phenotype affects only specific neuronal subpopulations; therefore, the effect of the drugs tested can be over- or under-interpreted. Another issue linked to heterogeneous neuronal populations is the reproducibility among different populations. Varying subpopulations of neurons with different cellular disease phenotypes is another challenge since such subpopulations can vary significantly between different experiments and influence functional readouts. Another issue to take into consideration is that epilepsy is a complex neurological disorder, where excitatory and inhibitory neuronal communication is disturbed. The ideal case would be to generate a mixed population with both types of neurons, ideally with glial contributions to investigate the activities of neurons in interaction with each other or with glial cells, which is a more in vivo-like condition[66], but this would also be more challenging in terms of the interpretation of results and drug efficacy. The most in vivo-like situation would be comparative studies in vascularized cerebral organoids, but this line of research, which is described below, is still in its infancy and not applicable for large-scale drug tests.
iPSC-derived brain organoids in neurodevelopment and epilepsy
Epilepsy is a complex brain disorder with excitatory and inhibitory neuron involvement, in which neural network activity is further dependent on neurotransmitter recycling via astrocytes. Therefore, even though 2D models are useful in electrophysiological measurements and drug screening, they have limitations when it comes to investigation of more complex interactions. To imitate the natural physiological conditions, architecture and neurodevelopmental features of the brain, differentiated neurons can be organized into 3D cerebral organoids[67].
A more sophisticated model is the 3D cerebral organoid generated using intrinsic patterning, which can yield semi-organized organoids containing neurons from different parts of the brain[22,68]. For example hiPSC-derived cerebral organoids show primitive cortical organization, dorsal forebrain-like progenitor zones and expression of markers specific for the hippocampus and forebrain. Cerebral organoids are useful tools to replicate neuronal differentiation and assess the neuronal subtype composition of specific brain regions. Moreover, they are widely used to investigate neural migration and establishment of connective neural circuits reflecting brain development. These processes are essential for neurodevelopmental disorders including DEEs, which are very challenging to capture and investigate in 2D in vitro models or in in vivo models, which often times show species-specific differences.
A successful example of the application of 3D cerebral organoids in neurodevelopmental disorders is autism spectrum disorder, where cerebral organoids have revealed an accelerated cell cycle, increased number of synapses and an overrepresentation of GABAergic inhibitory neurons caused by overexpression of FOXG1[69]. Another disorder that was modeled with cerebral organoids is Miller-Dieker syndrome (MDS). MDS is caused by a deletion at 17p13.3 and characterized by severe intellectual disability, intractable epilepsy and lissencephaly. MDS cerebral organoids revealed a mitotic defect in outer radial glia, which are critical for human neocortical expansion[70]. These examples provide evidence that organoids can be very useful in understanding the pathology behind brain disorders, but the usefulness of organoids in large-scale drug efficacy tests is still challenging due to the variability of neuronal subtypes, organoid size and limited penetration of compounds into organoids due to the absence of vascularization.
Currently, some of the most promising research studies combine the fusion of cortical organoids enriched in glutamatergic neurons and sub-pallium organoids enriched in GABAergic neurons. These fused organoids show the development of GABAergic neural progenitors and their integration in glutamatergic organoids. Most importantly, in this system, GABAergic interneurons can functionally integrate with glutamatergic neurons to form an electrophysiologically active network[71,72].
A pioneer study in the field of organoids demonstrated that organoids generated from patients with primary microcephaly showed morphological and cellular defects similar to those observed in the postmortem brains of patients with microcephaly. These defects included abnormal migration and proliferation of neural progenitors, which resulted in smaller organoids compared to controls, reflecting the in vivo situation in microcephaly[73]. Even though recent advancements in organoid technologies have made it possible to standardize organoid growth[74], there are still size limitations implemented due to the development of a necrotic core in larger sized organoids. Those necrotic cores are caused by a switch from a proliferative neural progenitors to terminally differentiated neurons and impaired diffusion of oxygen and nutrients to the core along with hampered metabolite exchange. Those are now actively addressed via, for example, bioprinting attempts where endothelial cells are introduced via extrusion or droplet deposition of cells suspended in biocompatible gel in a patterned manner, sacrificial networks where specific matrices are used to seed neural progenitors and endothelial cells in the generated tubular structures, or more classical approaches of growing cerebral organoids in co-culture with endothelial cells, which will cause angiogenic sprouting from the endothelial cells and infiltration of the cerebral organoids[75]. Even though these are significantly increasing the complexity of working with cerebral organoids, vascularization would be a leap forward to generate more in vivo-like conditions. In the case of epilepsy studies, the functional analysis of changes in excitability is crucial. Here, major improvements have been made in applying optogenetics and MEA electrophysiology. Furthermore, next-generation imaging technologies can monitor neural activities in live imaging during the high throughput drug screening of antiepileptic drugs[24,76]. Despite the mentioned advantages, organoids are also associated with disadvantages including being time-consuming and having low reproducibility between experiments, and compound screens are extremely challenging in this system.
Consequently, all in vitro models need to be substantiated by relevant in vivo models to understand complex pharmacological processes and disease phenotypes. Several preclinical animal models for epilepsy have been used to develop potential treatment strategies. Unfortunately, these models have failed to translate into successful clinical trials. This is in part due to the critical differences in gene expression, protein function and network activities between animal models and humans[77]. These failures clearly indicate that the hiPSC models are superior in relation to drug development strategies. Nevertheless, one should not exclude in vivo models, since they are still highly relevant to assessing the bioactivity of compounds and the ability of those compounds to pass the blood-brain-barrier, which are essential to investigate before embarking on human clinical trials. The various experimental models and their unique properties are summarized in[22].
Conclusion
2D hiPSC-derived neuronal systems generated from epilepsy patients and precise gene-editing techniques via CRISPR/Cas9 provide powerful platforms to investigate functional consequences of gene variants and to perform drug screening. Furthermore, cerebral organoids allow for more complex investigations of different neural populations and 3D neural circuits. Even though the 2D model system lacks spatial and regional complexity it is more suitable for understanding cellular pathologies. The 3D model is advantageous to understand developmental connectivity issues between different neurons and the effect on cortical layer development. On the down side, electrophysiological recordings and cell viability in the core of such cerebral organoids is still challenging; since vascularization is an important factor in organoid function and survival, various efforts are being made to improve this. Taken together, both 2D and 3D hiPSC models from epilepsy patients are superior to cancer-based human cell models, non-human cell models or frog oocytes to understand the underlying pathology. Lastly, hiPSC-derived neural progenitors have the future prospect of autologous stem cell therapy, combined with precision gene editing to repair the pathogenic variants.
Declarations
Authors’ contributionsContributed to conception and study design: Mohammadi NA, Møller RS
Drafted and revised the manuscript and figures with feedback from all of the other authors: Mohammadi NA
Contributed to generation of Figure 2 and drafting the table: Freude K, Haukedal H
Zeynep Tümer, Rikke S. Møller and Kristine Freude contributed to critical review of the manuscript: Freude K, Tümer Z, Møller RS
Availability of data and materialsWhat we have of information and data are from already published artikels, meaning that they can be available to readers.
Financial support and sponsorshipThe study was supported by the Novo Nordisk Foundation (NNF19OC0058399).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. England MJ, Liverman CT, Schultz AM, Strawbridge LM. Summary: a reprint from epilepsy across the spectrum: promoting health and understanding. Epilepsy Curr 2012;12:245-53.
2. Epilepsy. 2019. Available from: https://www.who.int/news-room/fact-sheets/detail/epilepsy [Last accessed on 9 Jun 2020].
3. Fisher RS, van Emde BW, Warren B, Christian E, Pierre G, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005;46:470-2.
4. Stafstrom CE, Carmant L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med 2015;5:a022426.
5. Mormann F, Kreuz T, Andrzejak RG, David P, Lehnertz K, et al. Epileptic seizures are preceded by a decrease in synchronization. Epilepsy Res 2003;53:173-85.
6. Moller RS, Hammer TB, Rubboli G, Lemke JR, Johannesen KM, et al. From next-generation sequencing to targeted treatment of non-acquired epilepsies. Expert Rev Mol Diagn 2019;19:217-28.
7. Brunklaus A, Du J, Steckler F, Ghanty II, Johannesen KM, et al. Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 2020;61:387-99.
8. Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017;140:1316-36.
9. Moller RS, Larsen LHG, Johannesen KM, Talvik I, Talvik T, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol 2016;7:210-9.
10. Gardella E, Moller RS. Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia 2019;60:S77-85.
12. Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, et al. Ion channels in genetic epilepsy: from genes and mechanisms to disease-targeted therapies. Pharmacol Rev 2018;70:142-73.
13. Milligan CJ, Li M, Gazina EV, Heron SE, Nair U, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol 2014;75:581-90.
14. Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 2017;140:2337-54.
15. Jiang X, Raju PK, D’Avanzo N, Lachance M, Pepin J, et al. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia 2019;60:1881-94.
16. Strehlow V, Heyne HO, Vlaskamp DRW, Marwick FFM, Rudolf G, et al. GRIN2A-related disorders: genotype and functional consequence predict phenotype. Brain 2019;142:80-92.
17. Shao LR, Habela CW, Stafstrom CE. Pediatric epilepsy mechanisms: expanding the paradigm of excitation/inhibition imbalance. Children (Basel) 2019;6:23.
18. Marafiga JR, Pasquetti MV, Calcagnotto ME. GABAergic interneurons in epilepsy: more than a simple change in inhibition. Epilepsy Behav 2020;106935.
19. Perucca P, Mula M. Antiepileptic drug effects on mood and behavior: molecular targets. Epilepsy Behav 2013;26:440-9.
21. Heyne HO, Singh T, Stamberger H, Jamra RA, Caglayan H, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet 2018;50:1048-53.
22. Maljevic S, Reid CA, Petrou S. Models for discovery of targeted therapy in genetic epileptic encephalopathies. J Neurochem 2017;143:30-48.
23. Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol 2009;87:41-57.
25. Barker BS, Ottolini M, Wagnon JL, Hollander RM, Meisler MH, et al. The SCN8A encephalopathy mutation p.Ile1327Val displays elevated sensitivity to the anticonvulsant phenytoin. Epilepsia 2016;57:1458-66.
26. Zeng SL, Sudlow LC, Berezin MY. Using Xenopus oocytes in neurological disease drug discovery. Expert Opinion on Drug Discovery 2020;15:39-52.
27. Quraishi IH, Stern S, Mangan KP, Zhang Y, Ali SR, et al. An epilepsy-associated KCNT1 mutation enhances excitability of human iPSC-derived neurons by increasing slack KNa currents. J Neurosci 2019;39:7438-49.
28. Grabole N, Zhang JD, Aigner S, Ruderisch N, Costa V, et al. Genomic analysis of the molecular neuropathology of tuberous sclerosis using a human stem cell model. Genome Med 2016;8:94.
29. Homan CC, Pederson S, To TH, Tan C, Piltz S, et al. PCDH19 regulation of neural progenitor cell differentiation suggests asynchrony of neurogenesis as a mechanism contributing to PCDH19 girls clustering epilepsy. Neurobiol Dis 2018;116:106-19.
30. Higurashi N, Uchida T, Lossin C, Misumi Y, Okada Y, et al. A human dravet syndrome model from patient induced pluripotent stem cells. Mol Brain 2013;6:19.
31. Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol 2013;74:128-39.
32. Stanurova J, Neureiter A, Hiber M, Kessler HO, Stolp K, et al. Angelman syndrome-derived neurons display late onset of paternal UBE3A silencing. Sci Rep 2016;6:30792.
33. Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, et al. Induced pluripotent stem cell models of the genomic imprinting disorders angelman and prader-willi syndromes. Proc Natl Acad Sci USA 2010;107:17668-73.
34. Bayat A, Hjalgrim H, Møller RS. The incidence of SCN1A-related dravet syndrome in denmark is 1:22,000: a population-based study from 2004 to 2009. Epilepsia 2015;56:36-9.
35. Maeda H, Chiyonobu T, Yoshida M, Yamashita S, Zuiki M, et al. Establishment of isogenic iPSCs from an individual with SCN1A mutation mosaicism as a model for investigating neurocognitive impairment in dravet syndrome. J Hum Genet 2016;61:565-9.
36. Fan J, Thalody G, Kwagh J, Burnett E, Shi H, et al. Assessing seizure liability using multi-electrode arrays (MEA). Toxicol In Vitro 2019;55:93-100.
37. Liu J, Sternberg AR, Ghiasvand S, Berdichevsky Y. Epilepsy-on-a-chip system for antiepileptic drug discovery. IEEE Trans Biomed Eng 2019;66:1231-41.
38. Odawara A, Matsuda N, Ishibashi Y, Yokoi R, Suzuki I. Toxicological evaluation of convulsant and anticonvulsant drugs in human induced pluripotent stem cell-derived cortical neuronal networks using an MEA system. Sci Rep 2018;8:10416.
39. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663-76.
40. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861-72.
41. Haase A, Olmer R, Schwanke K, Wunderlich S, Merkert S, et al. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 2009;5:434-41.
42. Luo S, Gu X, Ma F, Liu C, Shen Y, et al. ZYZ451 protects cardiomyocytes from hypoxia-induced apoptosis via enhancing MnSOD and STAT3 interaction. Free Radic Biol Med 2016;92:1-14.
43. Malik N, Rao MS. A review of the methods for human iPSC derivation. Methods Mol Biol 2013;997:23-33.
44. Vlahos K, Sourris K, Mayberry R, McDonald P, Bruveris FF, et al. Generation of iPSC lines from peripheral blood mononuclear cells from 5 healthy adults. Stem Cell Res 2019;34:101380.
45. Kamath A, Ternes S, McGowan S, Moy AB. Virus-free and oncogene-free induced pluripotent stem cell reprogramming in cord blood and peripheral blood in patients with lung disease. Regen Med 2018;13:889-915.
46. Haase A, Gohring G, Martin U. Generation of non-transgenic iPS cells from human cord blood CD34(+) cells under animal component-free conditions. Stem Cell Res 2017;21:71-3.
47. Okumura T, Horie Y, Lai CY, Lin HT, Shoda H, et al. Robust and highly efficient hiPSC generation from patient non-mobilized peripheral blood-derived CD34+ cells using the auto-erasable Sendai virus vector. Stem Cell Res Ther 2019;10:185.
48. Simara P, Tesarova L, Rehakova D, Farkas S, Salingova B, et al. Reprogramming of adult peripheral blood cells into human induced pluripotent stem cells as a safe and accessible source of endothelial cells. Stem Cells Dev 2018;27:10-22.
49. Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 2010;7:20-4.
50. Merling RK, Sweeney CL, Choi U, Ravin SSD, Myers TG, et al. Transgene-free iPSCs generated from small volume peripheral blood nonmobilized CD34+ cells. Blood 2013;121:e98-107.
51. Bang JS, Choi NY, Lee M, Ko K, Lee HJ, et al. Optimization of episomal reprogramming for generation of human induced pluripotent stem cells from fibroblasts. Anim Cells Syst (Seoul) 2018;22:132-9.
52. Lim SJ, Ho SC, Mok PL, Tan KL, Ong AHK, et al. Induced pluripotent stem cells from human hair follicle keratinocytes as a potential source for in vitro hair follicle cloning. Peer J 2016;4:e2695.
53. Matsumura W, Fujita Y, Nakayama C, Shinkuma S, Suzuki S, et al. Establishment of integration-free induced pluripotent stem cells from human recessive dystrophic epidermolysis bullosa keratinocytes. J Dermatol Sci 2018;89:263-71.
54. Kimura H, Ouchi T, Shibata S, Amemiya T, Nagoshi N, et al. Stem cells purified from human induced pluripotent stem cell-derived neural crest-like cells promote peripheral nerve regeneration. Sci Rep 2018;8:10071.
55. Shi L, Cui Y, Luan J, Zhou X, Han J. Urine-derived induced pluripotent stem cells as a modeling tool to study rare human diseases. Intractable Rare Dis Res 2016;5:192-201.
56. Gaignerie A, Lefort N, Rousselle M, Forest-Choquet V, Flippe L, et al. Urine-derived cells provide a readily accessible cell type for feeder-free mRNA reprogramming. Sci Rep 2018;8:14363.
57. Li Y, Nguyen HV, Tsang SH. Skin biopsy and patient-specific stem cell lines. Methods Mol Biol 2016;1353:77-88.
58. Klein T, Günther K, Kwok CK, Edenhofer F, Üçeyler N. Generation of the human induced pluripotent stem cell line (UKWNLi001-A) from skin fibroblasts of a woman with fabry disease carrying the X-chromosomal heterozygous c.708G>C (W236C) missense mutation in exon 5 of the alpha-galactosidase-A gene. Stem Cell Res 2018;31:222-6.
59. Karumbayaram S, Lee P, Azghadi SF, Cooper AR, Patterson M, et al. From skin biopsy to neurons through a pluripotent intermediate under good manufacturing practice protocols. Stem Cells Transl Med 2012;1:36-43.
60. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816-21.
61. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014;346:1258096.
62. Zhang Y, Pak CH, Han Y, Ahlenius H, Zhang Z, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013;78:785-98.
63. Lee S, Lim W, Ryu HW, Jo D, Min JJ, et al. ZW800-1 for assessment of blood-brain barrier disruption in a photothrombotic stroke model. Int J Med Sci 2017;14:1430-5.
64. Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 2009;27:275-80.
65. Watabe T, Miyazono K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Res 2009;19:103-15.
66. Nadadhur AG, Melero JE, Meijer M, Schut D, Jacobs G, et al. Multi-level characterization of balanced inhibitory-excitatory cortical neuron network derived from human pluripotent stem cells. PLoS One 2017;12:e0178533.
67. Simkin D, Kiskinis E. Modeling pediatric epilepsy through iPSC-based technologies. Epilepsy Curr 2018;18:240-5.
68. Tidball AM, Neely MD, Chamberlin R, Aboud AA, Kumar KK, et al. Genomic instability associated with p53 knockdown in the generation of huntington’s disease human induced pluripotent stem cells. PLoS One 2016;11:e0150372.
69. Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 2015;162:375-90.
70. Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, et al. Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell 2017;20:435-49.e4.
71. Birey F, Andersen J, Makinson CD, Islam S, Wei W, et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017;545:54-9.
72. Sloan SA, Andersen J, Paşca AM, Birey F, Paşca SP. Generation and assembly of human brain region-specific three-dimensional cultures. Nat Protoc 2018;13:2062-85.
73. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, et al. Cerebral organoids model human brain development and microcephaly. Nature 2013;501:373-9.
74. Krefft O, Jabali A, Iefremova V, Koch P, Ladewig J. Generation of Standardized and Reproducible Forebrain-type cerebral organoids from human induced pluripotent stem cells. J Vis Exp 2018;131:56768.
76. Sterlini B, Fruscione F, Baldassari S, Benfenati F, Zara F, et al. Progress of induced pluripotent stem cell technologies to understand genetic epilepsy. Int J Mol Sci 2020;21:482.
77. Simonato M, French JA, Galanopoulou AS, O’Brien TJ. Issues for new antiepilepsy drug development. Curr Opin Neurol 2013;26:195-200.
78. Zhang Y, Schmid B, Nikolaisen NK, Rasmussen MA, Aldana BI, et al. Patient iPSC-derived neurons for disease modeling of frontotemporal dementia with mutation in CHMP2B. Stem Cell Reports 2017;8:648-58.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mohammadi NA, Freude K, Haukedal H, Tümer Z, Møller RS. Human induced pluripotent cells in personalized treatment of monogenic epilepsies. J Transl Genet Genom 2020;4:238-50. http://dx.doi.org/10.20517/jtgg.2020.29
AMA Style
Mohammadi NA, Freude K, Haukedal H, Tümer Z, Møller RS. Human induced pluripotent cells in personalized treatment of monogenic epilepsies. Journal of Translational Genetics and Genomics. 2020; 4(3): 238-50. http://dx.doi.org/10.20517/jtgg.2020.29
Chicago/Turabian Style
Mohammadi, Nazanin A., Kristine Freude, Henriette Haukedal, Zeynep Tümer, Rikke S. Møller. 2020. "Human induced pluripotent cells in personalized treatment of monogenic epilepsies" Journal of Translational Genetics and Genomics. 4, no.3: 238-50. http://dx.doi.org/10.20517/jtgg.2020.29
ACS Style
Mohammadi, NA.; Freude K.; Haukedal H.; Tümer Z.; Møller RS. Human induced pluripotent cells in personalized treatment of monogenic epilepsies. J. Transl. Genet. Genom. 2020, 4, 238-50. http://dx.doi.org/10.20517/jtgg.2020.29
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 18 clicks
Cite This Article 18 clicks

 Like This Article 13
likes
Like This Article 13
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.