
Animal models of autism: a perspective from autophagy mechanisms

Abstract
Autism spectrum disorder (ASD) is characterized by impairments in social interaction and the presence of stereotypy and restrictive behavior. The clinical heterogeneity of ASD makes it difficult to explain the mechanisms underlying the disease. In recent years, the association between autophagy and neuropsychiatric diseases has been investigated. In this review, we aimed elucidate the relationship between autism and autophagy mechanism in well-known autism relevant animal models. Autophagy is a cell-protective mechanism that allows cell survival in low nutrient conditions, often through the degradation of aging and damaged proteins and organelles. The target of rapamycin (TOR) complex is activated for the activation of autophagy. Apart from mTOR animal models, the valproic acid model is frequently used in autism studies. The coiled-coil and C2 domain containing 1A (CC2D1A) gene is one of the new candidate genes associated with ASD. In a recent study that used Cc2d1a knock-out mice, microtubule-associated protein 1A/1B-light chain 3 (LC3) and Beclin 1 expression levels were dysregulated in the hippocampus. It is thought that the impaired autophagy mechanism contributes to the etiology of ASD. These results showed that CC2D1A acts as a new biological pathway in autophagy. Choosing the right model is crucial for ASD studies, and further progress will be made as these results become available in the clinic. In particular, it is expected that further studies on CC2D1A will provide new information in this field.
Keywords
Characteristics and genetic background of autism spectrum disorders
Autism spectrum disorders (ASD) is a lifelong disorder with onset during childhood which is characterized by rigidity and ritualistic/repetitive patterns of interest disturbing various brain regions including prefrontal, hippocampal, cerebellar, and striatal and other midbrain regions[1]. In approximately half of the cases, ASD is associated with intellectual disability (ID), comorbidity with other conditions including epilepsy, attention-deficit hyperactivity disorder, anxiety, depression, tics, sleep disorders, and gastrointestinal problems[2-7]. ASD prevalence and diagnostic rates have risen significantly over the past two decades reaching 1.0%-2.5%, with a male to female ratio of 4 to 1[8,9]. The reasons for sex discrepancy are not yet clearly understood[8,9]. Some studies argue that the differential expressions of ASD between sexes may result in an underdiagnosis of females because male patients tend to have more external behaviors such as increased repetitive behavior or aggression while female patients show more internal behaviors including depression, anxiety, and other emotional symptoms[2,10]. Males and females show similar levels of depression in childhood, but those levels diverge in adolescence, becoming dramatically greater in females[11]. During adolescence, females of autism are associated with high comorbidity including tic and eating disorders, high incidence of suicide, and high rates of other medical problems[12]. Additionally, ASD is diagnosed later in life in girls than in boys and some studies have reported that ASD symptoms may be milder in females or are masked by the occurrence of other comorbidities, such as depression, ID, or anxiety. Girls are expected to be more verbal and social by society than boys, and they can be more skilled at hiding social deficits[13].
ASD was considered a complex disorder that includes the complex interaction of genetic, epigenetic, and environmental factors which may lead to the alteration of brain structures and functions[14-16]. Clinical heterogeneity is the characteristic of ASD cases, and this heterogeneity is present at different levels of analysis such as genetics, neural systems, cognition, development, and behavior, as well as in clinical features from response to treatment[17]. Despite the current technological advances, the etiology of autism is still unknown and great efforts are being made to understand the nature of autism and its associated molecular pathways[18]. Rett’s syndrome, fragile X syndrome, and tuberous sclerosis (TS) are classified as syndromic ASD. Patients with syndromic autism have chromosome structure abnormalities or mutations[16]. These syndromes have a unique single gene mutation which is linked to the synaptic protein called as mammalian target of rapamycin (mTOR)[19]. mTOR is a critical protein responsible for dendritic plasticity and cell survival. mTOR may be implicated in disrupted cell signaling in idiopathic ASD. A unique gene has not been identified to cause idiopathic autism. Several candidate genes have been identified for autism[20-22]. Idiopathic autism that is not associated with a syndrome is called classic autism. It is estimated that rare genetic variants are responsible for about 10%-30% ASD cases[16]. Autism has a strong genetic basis with a complex transmission model that is thought to be the result of at least 1000 interacting genes[23,24]. These genes are associated with pathways such as cell growth and proliferation, synaptic activity and organization, transcription regulation, ubiquitination, chromatin rearrangement, protein synthesis, and transcription factors[25][Figure 1]. Several autism susceptibility genes including NRXN1, NLGN3, NLGN4, GABRB3, SHANK2, SHANK3, SCN1A, and CNTNAP2 have been identified by sequencing technologies[1,4,15]. Dysregulation of transforming growth factor β, bone morphogenetic protein (BMP), WNT/β-catenin, fibroblast growth factor (FGF), sonic hedgehog (SHH), and retinoic acid (RA) signaling pathways have been implicated in the pathogenesis of ASD[1].
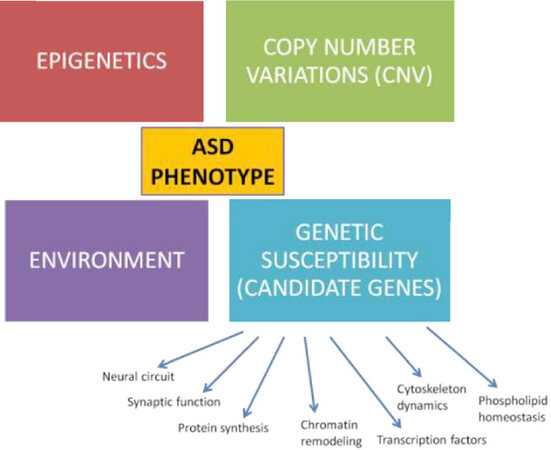
Figure 1. Genetic architecture of ASD. Genetic contributions to ASD can also be caused by direct or indirect effects on genes and proteins by environmental influences. ASD: autism spectrum disorder
Several alterations have been detected in both the macro and microscopic structures of the brain in autism. In most ASD cases, the cerebral cortex is qualitatively similar to typically developing subjects in its general organization[26-29]. Abnormalities of brain development may contribute to the autism pathology. Neuritogenesis may also represent the structural basis of autism pathology[30]. Abnormal functioning and changes of the cerebellum have been revealed in the postmortem brains of autism patients. There is a decrease in the number of Purkinje cells in the cerebellum[31]. Neuropsychiatric developmental disorders including schizophrenia and autism are associated with synapse development and abnormal dendritic spine formation[32]. Pathological events affecting the temporal lobe, especially the amygdala and the hippocampus, are thought to be related to the development of symptoms similar to autism[33]. Bilateral disorders in the cerebellum, thalamus, hippocampus, and amygdala regions were detected in autism[34].
Autophagy mechanism
Autophagy is a type of cell death mechanism that functions as an intracellular quality control system to maintain homeostasis by removing damaged proteins[35]. Autophagy is also a cell-protective mechanism that allows cell survival in low nutrient conditions and controls cell quality under stress conditions, often through degradation of aging and damaged proteins and organelles[36]. Autophagy is carried out by at least three different mechanisms, microautophagy, macroautophagy, and chaperone dependent autophagy. Macroautophagy occurs at the basic level in most cells, playing an important role in the breakdown of damaged organelles and proteins. Microautophagy is the event of lysis of the cytoplasm directly by the lysosome and digestion of the cytoplasm content within the lysosome with the lysosome membrane collapsing inward. And, chaperone-mediated autophagy selectively transfers proteins with KFERQ motifs to the lysosome membrane[37].
Autophagy is induced by starvation, oxygen deficiency, and various stress conditions. In these cases, the target of rapamycin (TOR) complex is activated for the initiation of autophagy. The TOR complex is the key protein that controls the cell’s energy metabolism, protein synthesis, and cell growth. It was first identified as the target molecule of rapamycin, an immunosuppressive agent developed for use against yeast in fungi. The isoform in mammals is known as mTOR, and suppression of this protein or silencing of the mTOR gene through various gene modifications provides stimulation of autophagy[38]. It is also known that the dysregulation of the autophagy mechanism is associated with human diseases such as cancer and neurodegeneration. Therefore, autophagy has attracted considerable interest in the biological sciences[39]. Several autophagic proteins control this pathway. BECN1 is a principal player in autophagy, triggering autophagosome formation[40]. LC3, serving as another important marker of autophagy, undergoes cleavage to become LC3-I and is then conjugated to phosphatidylethanolamine (PE), to produce LC3-II[41][Figure 2].
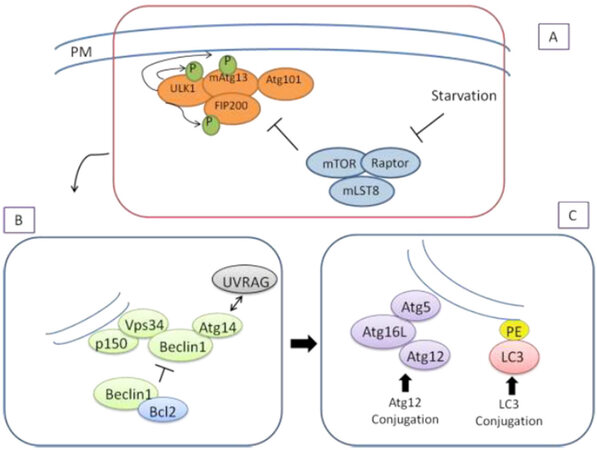
Figure 2. Molecular mechanism of autophagosome formation in mammalian autophagy process. Three major steps of the autophagy are figured out as initiation (A), nucleation (B) and elongation (C). PM: plasma membrane; PE: phosphatidylethanolamine
Neuronal autophagy is crucial for the interaction, signaling, and development of neurons, and the alteration of autophagy negatively affects the growth and function of neurons. Proper growth of axons and dendrites is important for neuronal balance. Organelles or proteins with impaired functions are normally degraded for structural plasticity during development. Recent studies have shown that genes associated with autophagy are very important in the development and maturation of dendrites, axons, and synapses[42]. ASD, as a neurodevelopmental disorder, may result in synaptic abnormalities. Autophagy and its related pathways are thought to be associated with the development of ASD, but the precise roles of neuronal autophagy in ASD were not fully understood[43,44]. Some studies have shown that activation of excess autophagy causes autism, while other studies have shown that deficiency of autophagy causes autism[24,29,45-48]. Recently, in the postmortem brains of patients with ASD, impaired autophagy, and hyperactive mTOR signaling were observed[45]. Autophagy is inhibited in mice with phosphatase and tensin homolog (PTEN) mutation and these mice exhibit autistic behaviors and abnormal neuronal development. This result suggests that the dysregulation of autophagy in individuals with ASD[49]. Mutation of the activity-dependent neuroprotector protein (ADNP) gene has also been identified as one of the causes of ASD[50]. ADNP is a gene encoding a transcription factor located on the long arm of chromosome 20 (20q13.13). ADNP syndrome is characterized by global developmental delays, ID, speech barriers, and motor dysfunctions[51]. Deletions in this chromosomal region cause ID[52]. ADNP controls the expression of genes during embryonic and postnatal development, including neurogenesis-crucial genes and BECN1[53]. Given the direct relationship between ADNP and MAP1-LC3B, as well as the connection of autism with eukaryotic initiation factor 4E (Eif4e) and its tight relationship with autophagy, ADNP is also inevitable to play a role in autism and autophagy[51,54]. ADNP knock-out mice resulted in embryonic death during the closure of the neural tube[53]. ADNP +/- male mice showed cognitive deficiencies in behavior tests[55].
Animal models on autophagy: a perspective from autism
An ideal animal model of a human disorder should present some characteristics features of the disease. The model should resemble the symptoms of human disorder and can be genetically modified by specific stimulation. Both the models and human patients should respond similarly to certain treatments[16]. Animal models can provide an advantage over human studies by allowing controlled testing of the effects of specific disease-causing factors on synaptic function and behavioral outcomes[56].
Many of the known genetic variations which contribute to the risk of ASD affect the expression of proteins that have roles in the chromatin remodeling or function, formation, and maintenance of synapses[57]. Deletions of such genes in animals can cause a behavioral phenotype reminiscent of ASD with impairments in communication, social interaction, and repetitive behaviors[56]. Knock-out models that include monogenic ASD genes are NRXN1, MECP2, NLGN3, SHANK3, NLGN4, FMR1, and TSC1/2[45,58-64]. Although there are few neuropathological data available in some models, there is growing interest of carrying specific ASD genes. The emerging risk genes are as follows: CHD8, SCN2A, SYNGAP1, DSCAM, and TBR1[65-69]. In the literature, there are syndromic animal models (i.e., Rett Syndrome, Prader-Willi, and Angelman Syndromes, fragile X, and Tuberous Sclerosis Complex) and environmental animal models [i.e., valproic acid (VPA), maternal autoantibodies, and maternal immune activation] that can be generated. In this review, the most studied mTOR related models and VPA models related to autophagy are discussed. Also, we explain the developments in the coiled-coil and C2 domain containing 1A (CC2D1A) animal models, which are not well-known in the literature, through the autophagy mechanism.
mTOR related models
The correct protein level is provided by the delicate balance between protein synthesis and breakdown. High synaptic protein levels can occur as a result of high translation and/or accumulation of damaged protein. In neurons, mTORC1 takes the role of braking autophagy. It is strategically positioned in the presynaptic and postsynaptic regions. Under rich nutrient conditions, the target of mTORC1 is Unc-51-like autophagy-activating kinase 1 (ULK-1) which phosphorylates in Ser757, the anti-autophagy region[70]. In this case, ULK-1 moves away from the AMP kinase (AMPK) and autophagy initiation is stopped. On the contrary, in the starvation conditions, AMPK phosphorylates and activates ULK-1 in Ser317 which mediates the phosphorylation and activation of Beclin-1 in Ser14, an important step in the “nucleation phase” of autophagy[71]. Beclin-1 promotes the lipidization of LC3-I to achieve the lipid form LC3-II which enables membrane elongation and formation of autophagosomes[72]. With lipidation, LC3-II is localized to the phagophore membrane and mediates the formation of mature LC3-II autophagosomes, limiting membrane elongation and cargo[73]. Neuronal autophagy has a key role in protein balance and is an important regulator of memory formation, synaptic plasticity, and structural remodeling[73,74].
A possible scenario is that over-activated mTOR from hippocampal neurons from fragile X mice results in decreased autophagy and accumulation of a particular group of synaptic proteins. Neurons from Fmr-1 knock-out mice showed the accumulation of ubiquitin-protein aggregates localized by p62, and autophagy was impaired in Fmr-1 knock-out neurons. This findings in this study showed that autophagy and protein degradation decreased in hippocampal neurons of this model. Excess mTOR activity is causally associated with decreased autophagy causing spinal defects, impaired cognition, and exaggerated synaptic plasticity in Fmr1- knock-out mice[75].
Interestingly, a recent study found that increased activity of mTOR and MAPK pathways in the peripheral blood samples of idiopathic ASD patients. ERK1-2, rpS6, p-eIF4E, and p-MNK1 (components of the mTOR and MAPK signaling pathways) showed a significant increase in patients with ASD compared to controls. TSC1, rpS6, p-eIF4E, and p-MNK1 protein expression discriminated patients according to their clinical severity[76]. Overactive mTOR signals suppress autophagy in the brains of tuberous sclerosis complex (TSC) 1+/- and TSC2+/- mice during postnatal development and decreased autophagy causing an imbalance in the pruning of the spines in the cortical layer V pyramidal neurons. Dysregulation of autophagy and its association with impaired spinal pruning may be more common in at least one subset of ASD in Tsc mutant mice[45]. TS is a genetic disease caused by mutations in the TSC1 or TSC2 genes and mTOR activity is regulated negatively with these genes[77]. TSC1+/- mice showed impairments in social interactions and hippocampus-dependent contextual fear conditioning[78]. TSC2 mutations created learning and memory deficits and contextual fear conditioning[79]. Mice carrying a dominant-negative TSC2 mutation had reduced social interactions and preference for social innovations[80].
Human with mutations in the PTEN gene tend to develop ASD, macrocephaly, seizures, and ID. It is thought that neurological symptoms associated with the loss of PTEN and other “mTORopathies” are caused by hyperactivation of mTORC1-mediated protein synthesis. One study with Pten knock-out mice revealed that rapamycin-mediated inhibition of MTORC1 activity increased behavioral and neurophysiological abnormalities and showed a reduction in brain size. The group also found that genetic deletion of mTORC2 activity suppressed seizures, recovered ASD-like behavior and long-term memory, and normalized metabolic changes of Pten knock-out mice. They found that reducing mTORC2 rescued behavioral and neurophysiological abnormalities[81]. The contactin associated protein 2 (CNTNAP2) is the first widely studied autism susceptibility gene. CNTNAP2 knock-out mice show core ASD-like phenotypes. By RNA sequencing of CNTNAP2 knock-out mouse, hyperactive Akt-mTOR signals were detected in the hippocampus. After treatment with mTOR inhibitor rapamycin or Akt inhibitor LY294002, it was reported that the social deficit was recovered in mice but had no effect on hyperactivity and recurrent/restricted behavior. Additionally, the effect of rapamycin and LY294002 on social behavior is reversible. Thus, the hyperactive Akt-mTOR signaling pathway has been identified as a therapeutic target for abnormal social behavior in patients with CNTNAP2 dysfunction[82].
According to latest study by Lieberman et al.[83], autophagy is downregulated during postnatal development following the upregulation of mTOR activity in the mice striatum. In the same study, a VPA model has been also conducted; autophagy is specifically reduced. They concluded that the impairment of autophagy is accompanied by impairments in synaptic transmission and social behavior in the late postnatal development in this mouse model[83]. In the TRIM32-/- model, the authors generated impaired GABAergic interneurons and leading autism relevant behaviors in mice, concomitant with an increased autophagy mechanism. Therefore, they suggested that theTRIM32-/- mouse is a novel autism mouse model[84].
VPA models
VPA is commonly used as an antiepileptic drug. Clinical studies have shown that exposure to VPA in utero is associated with cognitive deficits, birth defects, and an increased risk of ASD[85]. Clinical evidence shows that there is a link between VPA exposure and both cognitive abnormalities and autism. Animal studies in recent years have investigated anatomical, behavioral, molecular, and physiological changes due to in utero VPA exposure. The behavioral tests revealed that VPA exposure causes autistic-like behavior in offspring; these include social behavioral deficits, increased repetitive behavior, and communication deficits in rodents[86]. In the embryonic period, a single intraperitoneal VPA injection to 12.5-day-old female rats causes autism relevant symptoms in their offspring, and the brain structures and biomarker levels of the offspring are similar to those in autistic patients[87,88]. The VPA model is often used in ASD studies[89-91]. mTOR, the main marker of cellular metabolism, cell growth, and autophagy, has been reported to contribute to ASD development via the IGF1/PI3K/AKT/mTOR pathway[92]. Accordingly, it was assumed that autophagy will increase or decrease through PI3K/AKT/mTOR, thereby contributing to the development of ASD. Based on this assumption, VPA was given to rats and it was observed that the rats had autistic-like behaviors. Zhang et al.[46] rats were given VPA and autistic-like behaviors were observed. Various autophagic markers were examined in the hippocampus of these rats by the immunohistochemical method and mTOR inhibition was found to increase PI3K/AKT/mTOR-mediated autophagic activity.
Sphingosine 1-phosphate (S1P) is abundant in the brain tissue and plays an important role in brain development, regulation of neuronal differentiation, proliferation, survival, and apoptosis[93]. There is growing evidence that abnormal S1P levels are associated with the pathogenesis of schizophrenia, Alzheimer’s disease, and anxiety disorder[94]. In a study that investigated the pathogenesis of S1P in autism, a VPA rat model was used to evaluate S1P expression levels in the serum and brain tissue. Expression of autophagic proteins Beclin-1, LC3-II, and P62 were investigated in rats exposed to VPA. The investigators in the study found that increased S1P may be associated with decreased autophagy in this model[95]. A study was carried out to illuminate the link between the Notch signaling pathway and the pathogenesis of autism. They also investigated whether 3,5-difluorophenacetyl-L-alanyl-S-phenylglycine-2-butyl ester (Dapt) caused autism relevant behavior in the VPA rat model by regulating autophagy and affecting the morphology of dendritic spines. Autophagy-dependent proteins LC3B, Becn1, and phospho-p62 were inhibited by Dapt in rat VPA model in the hippocampus, cerebellum, and prefrontal cortex. The Notch signaling pathway participates in the ASD pathogenesis by affecting dendritic spine growth and regulating autophagy[96].
CC2D1A models for autism
The CC2D1A (coiled-coil and C2 domain-containing 1A) gene is one of the new candidate genes associated with autism[4]. In humans, the CC2D1A gene mutation was first associated with autosomal recessive nonsyndromic intellectual disability[97]. This means that the CC2D1A gene has a function in the central nervous system. Cc2d1a knock-out mouse model has been shown that regulates multiple intracellular signaling pathways, involved in neuronal differentiation and brain development, activators of Protein Kinase B (PKB), and NF-κB[98,99]. CC2D1A protein consists of a COOH-terminal C2 domain and NH2-terminal domains. The CC2D1A gene family consists of two homologous units, CC2D1A and CC2D1Bfor both humans and mice. The CC2D1A protein has two main isoforms containing 950 and 388 amino acids. The shorter isoform of the protein does not include exons 14 to 16, which are deleted in patients with ASD. Therefore, only the long protein isoform plays a central role in disease pathogenesis in the patients. The most conserved motif is a C2 domain and the other motif, DM14, is unique to the CC2D1A protein family, but its role is unknown. The C2 motif is located at the C-terminus of CC2D1A at positions 661-762, and is found in proteins that function in calcium-dependent phospholipid binding, where the C2 domain itself participates in the binding pocket of the Ca2+ cation. The DM14 motif repeats four times in the human CC2D1A sequence but only three times in the Caenorhabditis elegans orthologue sequence. The DM14 motif also appears only in the long isoform of CC2D1A, while the C2 domain is present in both isoforms, suggesting that it probably plays a central role in its protein activity[100-103]. The CC2D1A gene functions to bind to DNA and suppress transcription of serotonin receptor 1 A (HTR1A). The physiological role of the CC2D1A gene is not fully known. In a study, CC2D1A knock-out mice were produced to examine this role. Mice with the homozygous CC2D1A gene mutation died immediately after birth due to their inability to breathe, while mice with the heterozygous mutation remained alive and productive. Therefore, since the CC2D1A gene is vital, the absence of this gene as in the homozygous case is fatal[104]. The absence of the CC2D1A gene primarily affects brain function. Studies with animal models have revealed that this gene has a role in the regulation of endosomal traffic and signaling pathways. Disruption of this gene affects numerous biochemical pathways including cAMP response element-binding protein (CREB), NF-κB, protein kinase A (PKA), protein kinase B (PKB/AKT), Notch, and BMP[100]. CC2D1A plays a role in the transcriptional regulation o both dopamine and serotonin receptors in the brain. All studies have shown that CC2D1A regulated critical pathways for cognitive functions with neuronal differentiation[102,103]. Although there are not many studies in this animal model, abnormalities were found in the brain in the studies performed. Developmental changes in neurons of Cc2d1a knockout mouse brains were demonstrated during synapse maturation and induced neurotransmitter release. Based on these findings, it is thought that the CC2D1A gene serves as a developmental regulator of synapse function[100]. CC2D1A knock-out or knock-down in vitro experiments showed a decrease in hippocampal neurons. Also, delays in synaptic maturation have been observed in cortical neurons. In these animal models, it has been determined that there is a lack of neuronal plasticity, spatial learning, and memory accompanying decreased socialization, hyperactivity, anxiety, and excessive self-care[101] and Cc2d1a has been shown to control synaptic maturation of excitatory neurons[100]. Further studies are needed to determine whether CC2D1A controls sex-specific circuit function. Conditional removal of CC2D1A from the dorsal raphe demonstrated increased anxiety and depression-like behavioral phenotypes which correlated with reduced serotonin levels and increased 5HT-1A autoreceptor in the raphe, in both males and females[103], suggesting that there may be regional specificity in the function of CC2D1A.
In vitro studies in hippocampal neurons and embryonic fibroblasts from CC2D1A knock-out mice showed that CC2D1A binds to phosphodiesterase 4D (PDE4D), an enzyme involved in cAMP degradation[105]. In a recent study, PDE4D activation and downstream signaling molecules were tested in the hippocampus of Cc2d1a knock-out mice. Cc2d1a knock-out male mice were hyperactive and show a deficit in spatial memory, which led to a reduction in cAMP response element-binding protein signaling but this finding has not been correlated with female mice. These findings showed that CC2D1A regulates cAMP intracellular signaling in the male-specific regions of the hippocampus[106]. In our recent study, we showed the dysregulation of autophagy with CC2D1A deficient mice in the hippocampus. We wanted to evaluate the severity of autism by creating two different groups and followed them over the next three generations. LC3 and Beclin gene and protein expression levels in the hippocampus tissues of male and female mice in both groups were examined. All of the animal groups were observed to be extremely aggressive and hyperactive. Overall decreases were observed in autophagy levels. In the literature, this was the first major study in the CC2D1A mouse model in which autism was associated with autophagy[48].
Conclusion
Genetic studies of autism have made surprising progress over the past 20 years. Our understanding of the genetic and epigenetic factors in ASD etiology and the interaction on the disease will be continued to improve with future studies and ongoing research results.
Animal models are used to study potential disorder mechanisms. The well-known causes of autism are commonly based on specific human genetic mutations; however, ASD pathogenesis is most likely shaped by a complex interaction between several genetic variants as well as environmental factors in humans. In the animal models, monogenic mutations can lead to milder phenotypes that might explain some of the differences observed in behavioral manifestations between ASD patients and animal models. Given the clinical heterogeneity of the ASD patients, it is controversial whether it is necessary or even possible to see all the human symptoms in rodent models. However, monogenic rodent models are a valuable resource for solving the cause-and-effect relationships of ASD since the majority of susceptibility genes appear to converge in shared biological pathways. Therefore, rodent models are important preclinical tools necessary to investigate the validation of pathophysiology, gene function, and therapeutic approaches in ASD[107].
Choosing the right model is of great importance for ASD studies and progress will be made in the reflection of the results to be obtained in the clinic. Especially, the studies to be done with CC2D1A models are expected to gain new information in this field.
Declarations
Authors’ contributionsIdea conception: Sener EF
Read, Literature research, manuscript writing and editing, adjusted and approved the final manuscript: Dana H, Tahtasakal R, Sener EF
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Kumar S, Reynolds K, Ji Y, Gu R, Rai S, et al. Impaired neurodevelopmental pathways in autism spectrum disorder: a review of signaling mechanisms and crosstalk. J NeurodevDisord 2019;11:10.
2. Woodbury-Smith M, Scherer SW. Progress in the genetics of autism spectrum disorder. Dev Med Child Neurol 2018;60:445-51.
3. Anagnostou E, Zwaigenbaum L, Szatmari P, Fombonne E, Fernandez BA, et al. Autism spectrum disorder: advances in evidence-based practice. CMAJ 2014;186:509-19.
4. Sener EF, Cikili Uytun M, Bayramov KK, Zararsiz G, Oztop DB, et al. The roles of CC2D1A and HTR1A gene expressions in autism spectrum disorders. Metab Brain Dis 2016;31:613-9.
5. Bölte S, Girdler S, Marschik PB. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol Life Sci 2019;76:1275-97.
6. Bourgeron T. Current knowledge on the genetics of autism and propositions for future research. C R Biol 2016;339:300-7.
7. Bangerter A, Chatterjee M, Manyakov NV, Ness S, Lewin D, et al. Relationship between sleep and behavior in autism spectrum disorder: exploring the impact of sleep variability. Front Neurosci 2020;14:211.
8. Pagan C, Goubran-Botros H, Delorme R, Benabou M, Lemière N, et al. Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci Rep 2017;7:2096.
9. Rylaarsdam L, Guemez-Gamboa A. Genetic causes and modifiers of autism spectrum disorder. Front Cell Neurosci 2019;13:385.
10. Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol 2013;26:146-53.
11. Nolen-Hoeksema S, Girgus JS. The emergence of gender differences in depression during adolescence. Psychol Bull 1994;115:424-43.
12. Rynkiewicz A, Łucka I. Autism spectrum disorder (ASD) in girls. Co-occurring psychopathology. Sex differences in clinical manifestation. Psychiat Pol 2018;52:629-39.
13. Mossa A, Manzini MC. Molecular causes of sex-specific deficits in rodent models of neurodevelopmental disorders. J Neurosci Res 2019; doi: 10.1002/jnr.24577.
14. Carmassi C, Palagini L, Caruso D, Masci I, Nobili L, et al. Systematic review of sleep disturbances and circadian sleep desynchronization in autism spectrum disorder: toward an integrative model of a self-reinforcing loop. Front Psychiatry 2019;10:366.
15. Sener EF, Canatan H, Ozkul Y. Recent advances in autism spectrum disorders: applications of whole exome sequencing technology. Psychiatry Investig 2016;13:255-64.
16. Zhang XC, Shu LQ, Zhao XS, Li XK. Autism spectrum disorders: autistic phenotypes and complicated mechanisms. World J Pediatr 2019;15:17-25.
17. Bernard Paulais MA, Mazetto C, Thiébaut E, Nassif MC, Costa Coelho De Souza MT, et al. Heterogeneities in cognitive and socio-emotional development in children with autism spectrum disorder and severe intellectual disability as a comorbidity. Front Psychiatry 2019;10:508.
18. Sener EF, Taheri S, Sahin MC, Korkmaz Bayramov K, Marasli MK, et al. Altered global mRNA expressions of pain and aggression related genes in the blood of children with autism spectrum disorders. J of Molecular Neuroscience 2019;67:89-96.
19. Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet 2011;20:1182-96.
20. Sciara AN, Beasley B, Crawford JD, Anderson EP, Carrasco T, et al. Neuroinflammatory gene expression alterations in anterior cingulate cortical white and gray matter of males with autism spectrum disorder. Autism Res 2020;13:870-84.
21. C Yuen RK, Merico D, Bookman M, L Howe J, Thiruvahindrapuram B, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 2017;20:602-11.
22. Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009;459:569-73.
23. Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 1995;25:63-77.
24. Nicolini C, Fahnestock M. The valproic acid-induced rodent model of autism. Exp Neurol 2018;299:217-27.
26. Bauman M, Kemper TL. Histoanatomic observations of the brain in early infantile autism. Neurology 1985;35:866-74.
27. Guerin P, Lyon G, Barthelemy C, Sostak E, Chevrollier V, et al. Neuropathological study of a case of autistic syndrome with severe mental retardation. Dev Med Child Neurol 1996;38:203-11.
28. Hutsler JJ, Love T, Zhang H. Histological and magnetic resonance imaging assessment of cortical layering and thickness in autism spectrum disorders. Biol Psychiatry 2007;61:449-57.
29. Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res 2010;1309:83-94.
30. Bakos J, Bacova Z, Grant SG, Castejon AM, Ostatnikova D. Are molecules involved in neuritogenesis and axon guidance related to autism pathogenesis? Neuromolecular Med 2015;17:297-304.
31. Siniscalco D, Sapone A, Cirillo A. Autism spectrum disorders: is mesenchymal stem cell personalized therapy the future? J Biomed Biotechnol 2012;2012:480289.
32. Schuster S, Rivalan M, Strauss U. NOMA-GAP/ARHGAP33 regulates synapse development and autistic-like behavior in the mouse. Mol Psychiatry 2015;20:1120-31.
33. Levitt JG, Blanton RE, Smalley S. Cortical sulcal maps in autism. Cereb Cortex Jul 2003;13:728-35.
34. Scott JA, Schumann CM, Goodlin-Jones BL, Amaral DG. A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Res 2009;2:246-57.
35. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014;15:81-94.
36. Costa L, Amaral C, Teixeira N, Correia-da-Silva G, Fonseca BM. Cannabinoid-induced autophagy: protective or death role? Prostaglandins Other Lipid Mediat 2016;122:54-63.
37. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Molecular Cancer 2015;14:48.
38. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011;18:571-80.
39. Yamamoto H, Fujioka Y, Suzuki SW, Noshiro D, Suzuki H, et al. The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev Cell 2016;38:86-99.
40. Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 2007;17:839-49.
41. Wu J, Dang Y, Su W, Liu C, Ma H, et al. Molecular cloning, and characterization of rat LC3A and LC3B-two novel markers of autophagosome. Biochem Biophys Res Commun 2006;339:437-42.
42. Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer’s disease. J Alzheimers Dis 2006;9:277-89.
43. Kim HJ, Cho MH, Shim WH. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry 2017;22:1576-84.
44. Lee KM, Hwang SK, Lee JA. Neuronal autophagy and neurodevelopmental disorders. Exp Neurobiol 2013;22:133-42.
45. Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, et al. Loss of mTOR dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014;83:1131-43.
46. Zhang J, Zhang JX, Zhang QL. PI3K/AKT/mTOR-mediated autophagy in the development of autism spectrum disorder. Brain Res Bull 2016;125:152-8.
47. Bowling H, Klann E. Shaping dendritic spines in autism spectrum disorder: mTORC1-dependent macroautophagy. Neuron 2014;83:994-6.
48. Dana H, Bayram KK, Delibaşı N, Tahtasakal R, Bayram R, et al. Disregulation of Autophagy in the Transgenerational Cc2d1a Mouse Model of Autism. Neuromolecular Med 2020;22:239-49.
49. Huber KM, Klann E, Costa-Mattioli M. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci 2015;35:13836-42.
50. Arnett AB, Rhoads CL, Hoekzema K, Turner TN, Gerdts J, et al. The autism spectrum phenotype in ADNP syndrome. Autism Res 2018;11:1300-10.
51. Sragovich S, Ziv Y, Vaisvaser S, Shomron N, Hendler T, et al. The autism mutatedADNP plays a key role in stress response. Transl Psychiatry 2019;9:235.
52. Amram N, Hacohen Kleiman G, Sragovich S, Malishkevich A, Katz J, et al. Sexual divergence in microtubule function: the novel intranasal microtubule targeting SKIP normalizes axonal transport and enhances memory. Mol Psychiatry 2016;21:1467-76.
53. Sragovich S, Merenlender Wagner A, Gozes I. ADNP plays a key role in autophagy: from autism to schizophrenia and Alzheimer’s disease. Bioessays 2017;39.
55. Malishkevich A, Amram N, Hacohen-Kleiman G, Magen I, Giladi E, et al. Activity dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer’s pathologies. Transl Psychiatry 2015;5:e501.
56. Varghese M, Keshav N, Jacot-Descombes S, Warda T, Wicinski B, et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol 2017;134:537-66.
57. De Rubeis S, Buxbaum JD. Recent advances in the genetics of autism spectrum disorder. Curr Neurol Neurosci Rep 2015;15:36.
58. Dudanova I, Tabuchi K, Rohlmann A, Südhof TC, Missler M. Deletion of alpha neurexins does not cause a major impairment of axonal pathfinding or synapse formation. J Comp Neurol 2007;502:261-74.
59. Hammer M, Krueger-Burg D, Tuffy LP, Cooper BH, Taschenberger H, et al. Perturbed hippocampal synaptic inhibition and γ-Oscillations in a Neuroligin-4 knockout mouse model of autism. Cell Rep 2015;13:516-23.
60. Chanda S, Aoto J, Lee SJ, Wernig M, Südhof TC. Pathogenic mechanism of an autism-associated neuroligin mutation involves altered AMPA-receptor trafficking. Mol Psychiatry 2016;21:169-77.
61. Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, et al. Autism-like deficits in Shank3-deficient mice are rescued by targeting actin regulators. Cell Rep 2015;11:1400-13.
62. Goffin D, Allen M, Zhang L, Amorim M, Wang IT, et al. Rett syndrome mutation MeCP2T158A disrupts DNA binding, protein stability, and ERP responses. Nat Neurosci 2012;15:274-83.
63. Grossman AW, Aldridge GM, Lee KJ, Zeman MK, Jun CS, et al. Developmental characteristics of dendritic spines in the dentate gyrus of Fmr1 knockout mice. Brain Res 2010;1355:221-7.
64. Feliciano DM, Quon JL, Su T, Taylor MM, Bordey A. Postnatal neurogenesis generates heterotopias, olfactory micronodules, and cortical infiltration following single cell Tsc1 deletion. Hum Mol Genet 2012;21:799-810.
65. Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, et al. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 2016;19:1477-88.
66. Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, et al. A gain-of function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 2001;102:307-17.
67. Aceti M, Creson TK, Vaissiere T, Rojas C, Huang WC, et al. Syngap1 haploinsufficiency damages a postnatal critical period of pyramidal cell structural maturation linked to cortical circuit assembly. Biol Psychiatry 2015;77:805-15.
68. Ka M, Chopra DA, Dravid SM, Kim WY. Essential roles for ARID1B in dendritic arborization and spine morphology of developing pyramidal neurons. J Neurosci 2016;36:2723-42.
69. Maynard KR, Stein E. DSCAM contributes to dendrite arborization and spine formation in the developing cerebral cortex. J Neurosci 2012;32:16637-50.
70. Easton CR, Dickey CW, Moen SP, Neuzil KE, Barger Z, et al. Distinct calcium signals in developing cortical interneurons persist despite disorganization of cortex by Tbr1 KO. Dev Neurobiol 2016;76:705-20.
71. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009;20:1992-2003.
72. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013;15:741-50.
73. Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012;11:709-30.
75. Shehata M, Matsumura H, Okubo-Suzuki R, Ohkawa N, Inokuchi K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci 2012;32:10413-22.
76. Yan J, Porch MW, Court-Vazquez B, Bennett MVL, Zukin RS. Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc Natl Acad Sci USA 2018;115:E9707-16.
77. Rosina E, Battan B, Siracusano M, Di Criscio L, Hollis F, et al. Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl Psychiatry 2019;9:50.
79. Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol 2007;62:648-55.
80. Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med 2008;14:843-8.
81. Chevere-Torres I, Maki JM, Santini E, Klann E. Impaired social interactions, and motor learning skills in tuberous sclerosis complex model mice expressing a dominant/negative form of tuberin. Neurobiol Dis 2012;45:156-64.
82. Chen CJ, Sgritta M, Mays J, Zhou H, Lucero R, et al. Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat Med 2019;25:1684-90.
83. Lieberman OJ, Cartocci V, Pigulevskiy I, Molinari M, Carbonell J, et al. mTOR suppresses macroautophagyduring striatal postnatal development and is hyperactive in mouse models of autism spectrum disorders. Front Cell Neurosci 2020;14:70.
84. Xing X, Zhang J, Wu K, Cao B, Li X, et al. Suppression of Akt-mTOR pathway rescued the social behavior in Cntnap2-deficient mice. Sci Rep 2019;9:3041.
85. Zhu JW, Zou MM, Li YF, Chen WJ, Liu JC, et al. Absence of TRIM32 leads to reduced GABAergic interneuron generation and autism-like behaviors in mice via suppressing mTOR signaling. Cereb Cortex 2020;30:3240-58.
86. Ornoy A, Weinstein-Fudim L, Ergaz Z. Prenatal factors associated with autism spectrum disorder (ASD). ReprodToxicol 2015;56:155-69.
87. Roullet FI, Lai JK, Foster JA. In utero exposure to valproic acid and autism--a current review of clinical and animal studies. Neurotoxicol Teratol 2013;36:47-56.
88. Schneider T, Przewłocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology 2005;30:80-9.
89. Yang EJ, Ahn S, Lee K, Mahmood U, Kim HS. Early behavioral abnormalities and perinatal alterations of PTEN/AKT pathway in valproic acid autism model mice. PLoS One 2016;11:e0153298.
90. Kim JW, Seung H, Kim KC, Gonzales ELT, Oh HA, et al. Agmatine rescues autistic behaviors in the valproic acid-induced animal model of autism. Neuropharmacology 2017;113:71-81.
91. Sheikh AM, Li X, Wen G, Tauqeer Z, Brown WT, et al. Cathepsin D, and apoptosis related proteins are elevated in the brain of autistic subjects. Neuroscience 2010;165:363-70.
92. Tian Y, Yabuki Y, Moriguchi S, Fukunaga K, Mao PJ, et al. Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J Pineal Res 2014;56:1-11.
93. Bozdagi O, Tavassoli T, Buxbaum JD. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol Autism 2013;4:9.
94. van Echten-Deckert G, Hagen-Euteneuer N, Karaca I, Walter J. Sphingosine-1-phosphate: boon and bane for the brain. Cell Physiol Biochem 2014;34:148-57.
95. Jang S, Kim D, Lee Y, Moon S, Oh S. Modulation of sphingosine 1-phosphate and tyrosine hydroxylase in the stress-induced anxiety. Neurochem Res 2011;36:258-67.
96. Wu H, Zhang Q, Gao J, Sun C, Wang J, et al. Modulation of sphingosine 1-phosphate (S1P) attenuates spatial learning and memory impairments in the valproic acid rat model of autism. Psychopharmacology (Berl) 2018;235:873-86.
97. Zhang Y, Xiang Z, Jia Y, He X, Wang L, et al. The Notch signaling pathway inhibitor Dapt alleviates autism-like behavior, autophagy, and dendritic spine density abnormalities in a valproic acid-induced animal model of autism. Prog Neuropsychopharmacol Biol Psychiatry 2019;94:109644.
98. Al-Tawashi A, Jung SY, Liu D, Su B, Qin J. Protein implicated in nonsyndromic mental retardation regulates protein kinase A (PKA) activity. J Biol Chem 2012;287:14644-58.
99. Manzini MC, Xiong L, Shaheen R, Tambunan DE, Costanzo SD, et al. CC2D1A regulates human intellectual and social function as well as NF-κB signaling homeostasis. Cell Rep 2014;8:647-55.
100. Zhao M, Raingo J, Chen ZJ, Kavalali ET. Cc2d1a, a C2 domain containing protein linked to nonsyndromic mental retardation, controls functional maturation of central synapses. J Neurophysiol 2011;105:1506-15.
101. Oaks AW, Zamarbide M, Tambunan DE, Santini E, Costanzo SD, et al. Cc2d1a loss of function disrupts functional and morphological development in forebrain neurons leading to cognitive and social deficits. Cereb Cortex 2017;27:1670-85.
102. Rogaeva A, Albert PR. The mental retardation gene CC2D1A/Freud-1 encodes a long isoform that binds conserved DNA elements to repress gene transcription. Eur J Neurosci 2007;26:965-74.
103. Vahid-Ansari F, Daigle M, Manzini MC, Tanaka KF, Hen R, et al. Abrogated freud-1/CC2D1A repression of 5-HT1Aautoreceptors induces fluoxetine-resistant anxiety/depression-like behavior. J Neurosci 2017;37:11967-78.
104. Basel-Vanagaite L, Attia R, Yahav M, Ferland RJ, Anteki L, et al. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J Med Genet 2006;43:203-10.
105. Al-Tawashi A, Gehring C. Phosphodiesterase activity is regulated by CC2D1A that is implicated in non-syndromic intellectual disability. Cell Commun Signal 2013;11:47.
106. Zamarbide M, Mossa A, Muñoz-Llancao P, Wilkinson MK, Pond HL, et al. Male specific cAMP signaling in the hippocampus controls spatial memory deficits in a mouse model of autism and intellectual disability. Biol Psychiatry 2019;85:760-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Dana H, Tahtasakal R, Sener EF. Animal models of autism: a perspective from autophagy mechanisms. J Transl Genet Genom 2020;4:251-62. http://dx.doi.org/10.20517/jtgg.2020.25
AMA Style
Dana H, Tahtasakal R, Sener EF. Animal models of autism: a perspective from autophagy mechanisms. Journal of Translational Genetics and Genomics. 2020; 4(3): 251-62. http://dx.doi.org/10.20517/jtgg.2020.25
Chicago/Turabian Style
Dana, Halime, Reyhan Tahtasakal, Elif Funda Sener. 2020. "Animal models of autism: a perspective from autophagy mechanisms" Journal of Translational Genetics and Genomics. 4, no.3: 251-62. http://dx.doi.org/10.20517/jtgg.2020.25
ACS Style
Dana, H.; Tahtasakal R.; Sener EF. Animal models of autism: a perspective from autophagy mechanisms. J. Transl. Genet. Genom. 2020, 4, 251-62. http://dx.doi.org/10.20517/jtgg.2020.25
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 30 clicks
Cite This Article 30 clicks

 Like This Article 24
likes
Like This Article 24
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.