
Chromodomain helicase DNA-binding proteins and neurodevelopmental disorders

Abstract
The significance of epigenomic regulation is now established in the etiology of neurodevelopmental disorders (NDDs). Epigenomic regulatory processes include chromatin remodeling as a major regulator of gene expression in development. Chromatin remodeling is an enzymatic process carried out by large multi-unit protein complexes, of which the chromodomain helicase DNA-binding proteins comprise one of four recognized major protein families, named the chromodomain helicase DNA-binding (CHD) family. There are nine CHD proteins (CHD1-9) encoded by nine correspondingly named CHD genes. Remarkably, five of the nine CHDs are already recognized to be causative of autosomal dominant syndromic NDD. In this review, we discuss the contribution of all CHDs to NDDs. And, we specifically focus on molecular studies involving CHD8 of which several have been recently published and scarcely reviewed. The widespread nature of downstream targeting for CHD8, as well as the finding of autosomal dominant disease for the majority of CHDs in general, implicates this family of chromatin remodelers as major players in NDD causation.
Keywords
Introduction
Introduction to epigenetics
The term epigenetics was initially introduced in 1942 as a means to describe hereditary (i.e., “genetic”) processes that were not explained by classical genetics[1]. While several epigenetic processes are now being defined both conceptually and mechanistically, the core idea remains the same, that is, gene function and regulation can be controlled by a non-DNA sequence environment. Currently three main epigenetic processes have gained prominence: DNA modification (mainly methylation), histone modification, and chromatin remodeling. A fourth process termed RNA interference[2] and associated RNA-based control mechanisms are also now coming into focus. For the purposes of this review, we will focus only on chromatin remodeling and specifically the role of the chromodomain protein family as chromatin remodelers. However, chromatin remodeling does not occur as a stand-alone process. On the contrary, it is closely linked to, and works in concert with, other epigenetic regulatory mechanisms. Therefore, we will define them briefly below.
Main types of epigenetic modifications
DNA modification
DNA modification refers to the process of adding/removing chemical moieties to/from the DNA sugar-phosphate backbone, usually at the 5 carbon position of cytosine. The best known of these modifications is DNA methylation; the addition or removal of methyl groups from the DNA backbone. It has been best characterized in the context of cytosine-guanine (CpG) dinucleotides, and is catalyzed by DNA methyltransferases[3]. Cytosine methylation within gene promoters has been typically associated with transcriptional repression: the presence of the methyl moiety alters the chemical stoichiometry and thus directly obstructs the binding of transcription factors, and/or may also locally induce a repressive chromatin structure that is nontolerant to transcription[4,5].
However, the relationship between DNA methylation and the regulation of gene expression is complex. For example, “CpG methylation” can be positioned at non-CpG sites, particularly in neuronal cells[6,7], and cytosine methylation may cause repression or activation of genes, depending on the context[5]. Finally, DNA methylation can also affect alternative splicing[8]. Therefore, this complexity requires careful interpretation. Recently, additional forms of DNA base modification have been recognized; DNA hydroxymethylation, carboxylation and formylation[9] are examples of other DNA modifications, albeit their roles are not yet well defined.
Histone modification
A second type of epigenetic control process is the modification of the tails of histone proteins by adding or removing chemical moieties. These moieties include, for example, methyl, acetyl, and sumoyl groups, among others. Modification of histones tails are more varied, in that they occur at several positions along the tails of different histones, and complicated, in that there can be more than one group added or removed at the same position, and their effect on gene function can also be more nuanced[10] compared to DNA modification. For example, methylation of the 4th and 27th lysine can occur as mono-, di- or trimethylation. However, H3K4me3 (trimethylation at the fourth lysine of histone 3) marks are mainly associated with activating gene promoters while H3K27me3 (trimethylation at the 27th lysine of histone 3) marks are associated with repressing regulatory regions[11,12].
Chromatin regulation
Importantly, both DNA methylation and histone modifications are known to interact to regulate chromatin structure and gene expression[13]. This process takes place involving the action of large multi-protein complexes called chromatin remodeling complexes[14]. They contain, in addition to positioning factors, key enzymes that directly control the regulation of gene expression, usually by catalyzing either DNA or histone modification reactions. The enzymes may be classified as: chromatin writers (e.g., histone methyltransferases and acetylases), erasers (e.g., histone demethylases and deacetylases), and readers (e.g., mainly chromodomain remodeling proteins)[15].
Chromatin readers include an important family of ATP-dependent DNA-binding proteins called chromodomain helicase DNA-binding (CHD) proteins, which are the focus of this review. These proteins are critical regulators of cellular processes such as proliferation, stem cell inactivity and cell fate determination[16]. Moreover, CHD proteins are associated with a wide variety of human diseases, though we focus here only on the role of CHD proteins and neurodevelopmental disease. We begin by detailing chromatin and chromatin remodeling complexes below.
Chromatin and chromatin remodelers
Chromatin
Chromatin which is composed of DNA and the histone protein scaffold it is wound around, exists either as condensed heterochromatin or as open euchromatin. Inside the nucleus, genomic DNA is tightly packaged by winding a 146-nucleotide section around a histone protein octamer to form a “nucleosome”. The histone octamer comprises two each of the histone proteins H2A, H2B, H3, H4 and also a linker histone H1. The core octamer histones contain amino acid “tails” that may be modified by chemical moieties[17].
Nucleosomes restrict DNA accessibility to the transcription machinery. However, they are dynamic; the DNA can become less or more tightly bound to the histone octamer, or the nucleosomes can become more or less closely positioned to each other. These structural changes are thought to be directly linked to gene transcription regulation, by making the DNA either more or less accessible to the transcription apparatus. Regulation of gene expression can be further fine-tuned by nucleosomes through incorporating histone variants and post-translational modifications to histone tails providing structural and hence functional complexity[18].
As the naked DNA must be accessed for DNA replication, transcription and repair, these processes all involve chromatin disruption and restoration, requiring dynamic changes in chromatin remolding. Thus structural alteration in chromatin structure facilitates downstream gene expression specific to cellular demand, and thereby holds significant importance in gene regulatory networks[19]. The dynamism of chromatin is a result of a symphony of changes that include remodeling of nucleosomes, modification of histones, presence/absence of non-histone DNA-binding proteins and non-coding RNAs. These alterations in chromatin structure are mostly carried out by chromatin remodelers[20].
Chromatin remodeling complexes
Chromatin remodelers are multiprotein complexes that catalyze nucleosome sliding (gliding of an octamer across the DNA) and histone variant exchange (changing the conformation of nucleosomal DNA)[20]. The remodeling enzymes can bring about change by ATP hydrolysis between the histone-DNA contact within the nucleosome. ATP-dependent chromatin remodelers utilize energy from ATP hydrolysis. They belong to the superfamily 2 helicases and share a conserved core ATPase[21]. ATP-dependent chromatin remodelers are further classified into 4 separate families: SWI/SNF (switch/sucrose-non-fermenting), ISWI (imitation switch), CHD (chromodomain helicase DNA-binding) and INO80 (inositol requiring 80). The distinction between them lies in the exclusive domains that reside adjacent to their ATPase domain. Briefly, SWI/SNF remodelers contain bromodomains, ISWI remodelers contain SANT-SLIDE modules, CHD remodelers contain tandem chromodomains, and the INO80 family contains HAS (helicase SANT) domains. These domains have distinctive roles in the regulation of ATPase activity, in recruiting remodelers, and for specific histone modifications[22]. Here, we will focus only on the CHD family.
CHD family
A number of proteins belonging to the chromodomain helicase DNA-binding family have been identified across phyla. In humans, nine CHDs are known (CHD 1 to 9), four are recognized in Drosophila (dCHD-1, dMi-2, Chd3 and Kismet) and one is known in yeast (yCHD1). They are well conserved, and distinguished by the presence of two chromatin organization modifier (“chromo”) domains located in the N-terminal region and two SNF-2-like ATP-dependent helicase domains positioned toward the center of the protein[23]. The chromodomains are capable of binding methylated DNA, RNA or histone tails[24] while the SNF-2 like ATP-dependent helicase domains confer enzymatic activity based upon ATP hydrolysis[25,26]. Thus, the CHD proteins may be summarized as effectors of chromatin conformational change via directional binding based upon methylation marks.
The nine human CHD proteins are further categorized into three groupings based upon their domain architecture as follows [Figure 1]: CHD1 and CHD2 are prototypical, not distinguished by other motifs but do contain a similar DNA-binding domain[25,26], and CHD3, CHD4 and CHD5 do not contain DNA-binding domains but do contain two plant homeodomain zinc-finger motifs (PHDs). PHDs are capable of recognizing post-translational modifications of histone tails[27], conferring sophisticated specificity for targeted chromatin remodeling. A significant example is the interaction between CHD3/4 and histone deacetylase that is part of the potent Nucleosome Remodeling Deacetylase (NuRD) transcriptional regulator complex[28]. The remaining CHDs, CHD6, CHD7, CHD8 and CHD9, are a diverse grouping with several additional domains, such as SANT, Brahma Kismet and conserved region, being recognized in their structure. The specific functions of these domains are not yet completely understood.
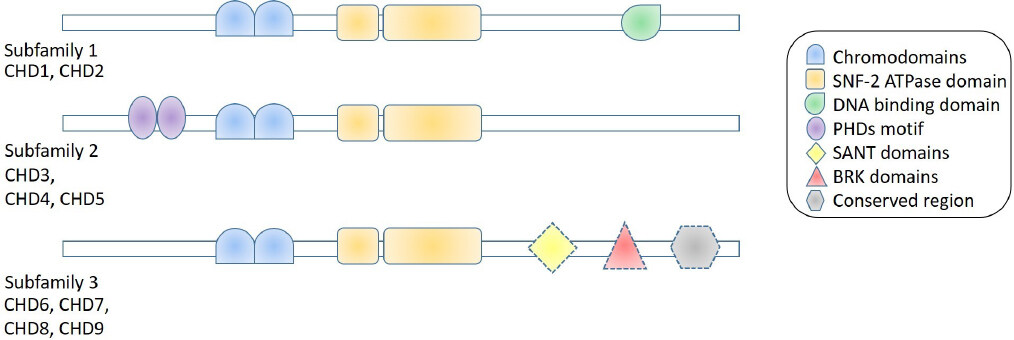
Figure 1. Schematic of chromodomain helicase DNA-binding (CHD) family protein structure. Subfamily 1 on the topmost row includes CHD1 and CHD2. They both contain the defining tandem two chromodomains and two SNF-2 ATPase domains - which all CHD proteins contain. They also contain a DNA-binding domain. Subfamily 2 on the middle row includes CHD3, CHD4 and CHD5, which in addition to definitive chromo and SNF-2 ATPase domains, also contain tandem PHD motifs. Subfamily 3 on the bottom row includes the other four CHD proteins (CHD6 to CHD9). Members of this group may have any combination of one or more SANT, BRK and conserved region domains (denoted by dashed lines)
Importantly, all CHDs are known to be ubiquitously expressed with the exception of CHD5, whose expression is confined to neuronal tissue[29]. A significant role in development for all CHDs has been demonstrated by stem cell research, where function in cell lineage and fate determination as well as cellular programming have been shown[16], indicating important roles for these proteins in human development and function. Here, we will focus on CHD roles in neurodevelopmental disorders (NDDs). However, we first begin by introducing NDDs and the contribution of epigenetic regulation in general to NDD causation.
Epigenetics in neurodevelopmental disease
Neurodevelopmental disorders
NDDs are a group of diseases that may be described as defects in the growth and development of the brain or nervous system. According to the current clinical designations for mental disorders by the American Psychiatric Association DSM-5 (Diagnostic and Statistical Manual, 5th edition, published in 2013), NDDs include: intellectual disability (ID), autism spectrum disorder (ASD), attention deficit/hyperactivity disorder, learning disorders, motor disorders and communication disorders. However, we note that from a molecular biology perspective, especially considering a common epigenetic etiology, other disorders such as bipolar disease and schizophrenia (SCZ) should be kept in mind as NDDs[30,31]. Among the NDDs, the most well studied are ID and ASD, which when taken together, are among the most common disorders worldwide, affecting 1%-3% of the global population[32,33].
Genetic sequence-based epigenetic causation in NDDs
The involvement of epigenetic processes in NDDs was first ascertained by the observation that several causative genes for monogenic NDD syndromes encode factors involved in epigenomic regulation, thereby directly implicating epigenomic deregulation as an etiology. We previously conducted a substantial review of NDDs caused by defects in genes that encode epigenetic factors[30], showing how such genetic defect-caused epigenomic deregulation leads to varied NDDs. Following the recognition that genetic mutations resulting in haploinsufficiency for proteins that are key epigenetic regulatory factors, or dominant negative forms thereof, can cause NDDs, focused studies probing the extent of causation found that epigenetic deregulation is indeed a significant etiology for NDDs[34], not limited to syndromic NDDs[35-37].
Environment-based epigenetic causes of NDDs
On a different track, twin studies showed that environmental exposures play a key role in NDD causation. Studying monozygotic twins displayed variation in behavior for psychiatric disorders, which was more prominent with age[38]. Currently, it is increasingly well recognized that adverse prenatal environments, such as exposure to maternal stress, viral infections, drugs, and toxins, can disrupt normal brain development and have lasting consequences on the structure and function of the brain[39,40]. Moreover, prenatal adversity can significantly increase the risk of developing mental disease in later life, including SCZ, depression, anxiety, and autism, among others[16,41,42]. Interestingly, maternal stress during pregnancy has also been associated with the increased risk of the development of many neuropsychiatric disorders in the offspring besides SCZ, including depression, autism, and anxiety[43].
The epigenome is highly susceptible to environmental exposures (e.g., maternal stress, toxins, drugs, pollutants) during early prenatal development, when extensive epigenetic reprogramming (resetting the human epigenome for naive pluripotency) and epigenetic programming (epigenetic alterations driving cellular differentiation) take place, to establish cell- and tissue-specific gene expression[44]. It is therefore hypothesized that interference with these processes during early embryogenesis can significantly impact early gene programming in the developing embryo[44,45]. In fact, there is a growing body of evidence suggesting that maternal exposures to epigenome regulation influencers such as “epi-diet” and “epi-drugs” - food substances and pharmacological compounds that influence the epigenome, respectively - are able to impact fetal outcome with respect to NDD[39].
Chromatin remodeling as important for plasticity during brain development and function
The ability of intrinsic and extrinsic factors that act via epigenetic mechanisms to effect NDD causation is explained by the fact that the brain is the most plastic of all organs. The developing and functioning brain must continuously adapt to external stimuli and respond to it. The response process, though not yet fully understood, almost certainly involves large-scale and fine-tuned chromatin changes that cause both structural and functional change at the molecular and cellular level during regulation of gene expression. Epigenetic regulatory processes are mechanistically capable of a range of fine-tuning of gene expression[46,47]. This in turn makes any change in the enzymes that lay out these processes a key susceptibility factor for NDD.
Epigenetic modifications of chromatin delivers dynamic regulation of gene expression via acetylation, phosphorylation, methylation, ubiquitination and sumoylation[48]. Thus, disturbance of this equilibrium in the brain or central nervous system leads to NDDs with complicated phenotypes[16,29]. While roles in NDD are found for all chromatin remodelers[49], we will focus here on that of the CHD family of SF-2 ATP-ase dependent remodelers.
CHDs and neurodevelopmental disease
CHDs known to cause NDDs
CHDs were first implicated in NDDs in the same way that epigenetics in general was recognized to be a significant etiological factor, by identifying causative genes for NDDs that happened to encode CHD proteins. The first to be so identified was CHD7, as the causative gene for CHARGE syndrome (CHARGE, an acronym for the common constellation of phenotypes presented - coloboma, heart defects, atresia choanae, retardation of growth, genital abnormalities, and ear abnormalities)[50]. After finding deletions in the gene in a small cohort of CHARGE patients by using a genome screen, a subsequent targeted sequencing of the CHD7 gene confirmed it to be the causative gene for CHARGE syndrome. However, while CHARGE syndrome is a developmental disorder, it is not considered primarily an NDD, since neurological phenotypes are not distinguishing. The next CHD gene identified to be disease-causing, and the first causative gene for an NDD, was CHD8, which was identified as one of two critical genes for a novel NDD syndrome first reported in 2007[51]. This finding was also the result of a genome screen in patients with ID. Subsequent work showed the gene to be the major causative factor for this syndrome, which we suggest be called Zahir Friedman syndrome (ZFS)[52].
Recently, with the widespread use of next-generation sequencing technologies, several more CHDs were identified as causative for NDD[23,53][Table 1]. Particularly, we note that syndromic NDDs caused by CHD1 (Pilarowski-Bjornsson syndrome- PILBOS)[54], CHD3 (Snijders Blok-Campeau syndrome -SNIBCPS)[55] and CHD4 (Sifrim-Hitz-Weiss syndrome - SIHIWES)[56], were identified in the past few years as a result of efforts to match genotype to phenotype using global repositories such as GeneMatcher. We anticipate that syndromic NDDs may be soon identified for CHD5, CHD6 and CHD9 as well, as genotype-phenotype correlations and genome screening for idiopathic patients with NDDs become ever more accessible globally.
NDDs identified that are caused by CHD gene defects
| CHD | Distinguishing features/comment | NDD that it is causative for |
|---|---|---|
| Subfamily 1 | ||
| CHD1 | Prototype - Two chromodomains and a SNF-2 ATPase domain
And DNA binding domain | Pilarowski-Bjornsson syndrome
Developmental Delay, Autism Spectrum Disorder, Seizures |
| CHD2 | Intellectual Disability, Epilepsy, Developmental Delay, Autism Spectrum Disorder | |
| Subfamily 2 | ||
| CHD3 | No DNA binding domain
Two PHDs motifs. CHD3, CHD4 and CHD5 form the core ATPase unit of the NuRD chromatin remodeling complex | Snijders Blok-Campeau syndrome.
Developmental Delay, Intellectual Disability, Seizures |
| CHD4 | Sifrim-Hitz-Weiss syndrome.
Intellectual Disability | |
| CHD5 | Yet unknown | |
| Subfamily 3 | ||
| CHD6 | Contain a variety of additional domains to the prototype
SANT domains, BRK domains, Conserved region domains | Yet unknown |
| CHD7 | CHARGE syndrome, Autism Spectrum Disorder | |
| CHD8 | Zahir Friedman syndrome, Autism Spectrum Disorder, Schizophrenia,
Intellectual Disability Developmental Delay Seizures | |
| CHD9 | Yet unknown |
In the past decade, there were also separate large-scale sequencing efforts to identify causative genes for ASD, which identified sequence mutations in CHD8 that can contribute to as much as 0.5% of ASD. While we posit that patients with sequence mutations in CHD8 also display ZFS[52], we do not rule out the possibility of a nuanced disease expression profile depending on whether the gene has undergone a deletion or sequence mutation. Interestingly, in a similar vein, the earliest reports of CHD2 as pathogenic were from microarray studies that found the gene involved in pathogenic deletions in patients with NDDs and epilepsy. Thereafter, targeted studies of large cohorts of patients with only epileptic encephalopathy (EE) or only ASD, identified mutations in CHD2, which resulted in CHD2 being termed an “EE gene” and an “ASD gene”[29,57]. It would be useful to compare the spectrum of phenotypes found for patients with CHD2 defects to understand whether epilepsy and ASD are distinguishing features of a broader syndromic NDD caused by defects in CHD2 (as we have shown for CHD8) to better understand CHD2 pathogenicity.
Below, we will compare and contrast phenotypic patterns for the NDD syndromes caused by CHD gene family defects.
Comparison of syndromic NDDs caused by CHD gene defects
Of the five known syndromes caused by CHDs, all except CHARGE syndrome (caused by CHD7), may be considered NDD syndromes primarily. CHARGE syndrome is often characterized as a major congenital anomaly condition, where nevertheless, patients may present with neurodevelopmental phenotypes such as ID[58]. The four syndromes, namely PILBOS caused by CHD1, SNIBCPS caused by CHD3, SIHIWES caused by CHD4, and ZFS caused by CHD8, are all autosomal dominant disorders, like CHARGE syndrome. While ZFS and SIHIWES are thought to occur due to haploinsufficiency, PILBOS is suggested to be caused by a dominant negative mechanism, while there is mixed evidence for SNIBCPS.
We have collated major reported phenotypes for these five syndromes in Table 2. While we compare them briefly below and in Table 2, we caution that an accurate and comprehensive comparison requires extensive curation of the variety of clinical assessment methods and situations covered by the plethora of publications reporting patients for all five syndromes. A task beyond the scope of this review; however, we make pertinent observations below.
Comparison of reported clinical presentation for CHD neurodevelopmental syndromes
| CHD1 Pilarowski-Bjornsson syndrome | CHD3 Snijders Blok-Campeau syndrome | CHD4 Sifrim-Hitz-Weiss syndrome | CHD7 CHARGE syndrome | CHD8 Zahir Friedman syndrome |
|---|---|---|---|---|
| Reference paper: (Pilarowski et al.[54] 2018) | Reference paper: (Snijders Blok et al.[55] 2018) | Reference paper: (Weiss et al.[81] 2016) | Reference paper: (Bergman et al.[82] 2011) | Reference paper: (Yasin et al.[52] 2019) |
| Total patients 5 | Total patients 35 | Total patients 5 | Total patients 280 patients from author’s own cohort only | Total patients 24. [51 patients are reported, but we only consider the 24 with detailed clinical data] |
| ID/DD 100% | ID/DD 100% | ID/DD 100% | DD 99%, ID 74% | ID/DD 100% |
| ASD or ASD like features 60% | Autism or autism-like features 29% | Hearing loss 80% | Coloboma 81% | ASD 86% |
| Speech Apraxia80% | Speech delay/disorder 100% | Macrocephaly 100% | Heart defects 76% | Speech defects 92% |
| Hypotonia 100% | Hypotonia 75% | Widely spaced eyes 100% | Choanal atresia 55% | Macrocephaly 86% |
| Macrocephaly 40% | Macrocephaly 58% | Dysmorphic ears 100% | Growth Retardation 37% | Behavioural defects 92% |
| Depressed midface 60% | Widened CSF spaces (MRI) 33% | Skeletal problems 100% | Genital anomalies 81% | Facial anomalies 100% |
| Translucent skin 60% | Neonatal feeding problems 31% | Brain morphological anomalies on MRI 100% | Ear anomalies 97% | Gastrointestinal defects 67% |
| Down-slanting palpebral fissures 60% | High, broad, and/or prominent forehead 85% | Heart defects 40% | Cranial nerve dysfunction 99% | Motor defects 67% |
| Seizures 60%-80% | Widely spaced eyes 77% | Feeding difficulties 82% | Sleep problems 54% |
Developmental delay/ID and/or ASD is a primary characteristic feature in patients with PILBOS, SNIBCPS, SIHIWES and ZFS[52,54-56]. Also, 27.5% of patients with CHARGE syndrome have been reported to have ASD[59], and ID is also reported in patients with CHARGE syndrome[60].
Notably, in addition to neurodevelopmental and neurocognitive phenotypes such as developmental delay and/or ID and/or ASD, patients from all four syndromes were reported to have macrocephaly. Speech delays are noted for PILBOS, SNIBCPS and ZFS patients. Of other characteristic phenotypes found in ZFS patients, hypotonia and some facial phenotypes such as hypertelorism and low-set ears in SNIBCPS, and skeletal phenotypes in SIHIWES are noted. Cardiac defects are seen in SIHIWES, as they are in CHARGE syndrome, and they have been reported in patients with ZFS. Finally, a characteristic facial gestalt is part of the presentation of SIHIWES, SNIBPCS and ZFS but not of the other two syndromes.
Due to the discovery of thus far five of the nine CHDs being disease-causing, there has been a flourishing of functional studies investigating pathophysiology in a number of model systems including animal and several human cellular types. Others have comprehensively reviewed these studies for several CHDs[61], for all CHDs[29], for CHD2[16], and for CHDs in stem cell function[16]. As these reviews are limited in their discussion of CHD8, we limit ourselves here to overviewing pertinent functional studies in CHD8 to fill the lacunae.
Role of CHD8 in NDD
There were only seven publications with the key word “CHD8” when it was first discovered as a causative gene for NDDs in 2007[51]. In just over the dozen years since then, a further 141 publications appear with “CHD8” as a key word, greatly enriching our understanding of the pathogenic and functional contribution of this gene. We note that several of these studies support an important role for CHD8 in cancer[62,63], which is beyond the scope of this paper. The burgeoning interest in CHD8 has no doubt been spurred by its reported causation of up to 0.5% of all ASD[64]. Extraordinarily, there are six different groups who have published findings from mouse models alone[65-70], as well as others who report functional studies with C. elegans[71] and Drosophila[72], and the first functional model study results were obtained with zebrafish[73]. Furthermore, a growing number of groups are exploring functional studies in human cellular models[74-77]. We will briefly overview the main findings from these model studies below.
Human cellular model studies
To our knowledge, the first human cellular model study was presented by Sugathan et al.[74] who used induced pluripotent stem cell-derived neural progenitor cells (NPCs) to examine CHD8 regulation in transcriptional networks. They knocked down CHD8 to single allele expression levels and then examined whole transcriptome and genome-wide ChIP-seq data to derive genome-wide impacts of the haploinsufficiency. They found that the expression of 1,756 genes was altered overall, with downregulated genes enriched for pathways involved in brain development. In addition, a total of 5,658 genes were shown to have possible CHD8-binding sites, and these were associated with transcriptional regulation and chromatin modification. These data indicate widespread downstream targeting.
A later study examining CHD8 knockdown in commercial human NPCs derived from neuroblastoma cells, discovered altered expression of both protein-coding and noncoding RNA genes, with a total of 1715 genes showing altered expression[77]. This cited study also found that the differentially expressed genes were enriched for neuronal development pathways and included known ASD candidate genes, similarly to Sugathan et al.[74].
A separate group generated iPSC-derived NPCs from a human donor using CRISPR/Cas-9 heterozygous gene disruption in CHD8[76]. They followed this by generating neurons and then brain organoids from the same source[75]. They observed 1,248 and 3,248 genes differentially expressed in NPCs and neurons, respectively, with an enrichment for genes involved in neuronal development and β-catenin/Wnt signaling, including known ASD and SCZ candidate genes[76]. Interestingly, they highlight the involvement of genes associated with brain volume among their differentially expressed genes, speculating a connection to the macrocephaly phenotype observed for patients with ZFS. The subsequent publication from this group of their findings for brain organoids showed a much lower number of differentially expressed genes, i.e., 559[75]. Nevertheless, neuronal development was once more enriched as was Wnt/β-catenin signaling. Of note from this study was the finding of a non-coding RNA, DLX6-AS1, as their topmost differentially expressed gene with a staggering +39 fold change in expression level[75], a gene gaining increasing importance in ASD pathophysiology[78].
We note that while each of the above studies followed rigorous established experimental protocols, there is a remarkable variation in the resulting differentially expressed gene datasets, with overall non-robust overlap, which could be the result of variation in both biological and bioinformatics experimental methods. However, in this respect, a meta-analysis conducted by running all the raw data from these studies through the same bioinformatics processing pipeline failed to eliminate or reduce the discordance in a meaningful manner[64]. In summary, the lack of any one or handful of consistent downstream target gene(s) for CHD8 from these studies is an observation that only further emphasizes that CHD8 indeed does have widespread downstream targeting, suggesting it is a possible master-regulator of transcription that displays both genomic and cellular context-dependent variation. It also underlines the importance of pathway-based data analyses when deciphering the results of gene expression studies for CHD8 deficiency models.
Studies in animal models
The earliest animal model for CHD8 was presented as a functional study in zebrafish that cemented the pathogenicity of the gene for macrocephaly[73]. However, the development of mouse models of Chd8+/- by several groups now dominates the characterization of haploinsufficiency of this gene in a complete organism, and significantly, from a mammalian system. While the sum characterizations from the mouse models offer a rich trove of information, we still note that all of the studies we are aware of[65-70], investigated the gene defect only as a major causative gene for ASD with the additional noted phenotype of macrocephaly, and not as one causing syndromic NDD. This bias must be kept in mind, as it greatly impacts the characterization results, since only ASD or related phenotypes were investigated. Nonetheless, observations from the mouse model studies offer key insights into the molecular physiology of Chd8 haploinsufficiency.
The first mouse model report in 2016 involved in utero-targeted cortical knockdown of Chd8[65]. Consistent with findings from the cellular model studies, a key role in the regulation of Wnt signaling (as a positive regulator) was observed. A distinct role in mammalian cortical development was proposed, acting via cell cycle regulation and PRC complex targeting. And behavioral deficits, in keeping with ASD equivalent phenotyping, were also noted[65]. In the same year, another group reported the characterization of a Chd8 heterozygous knockout mouse model[67]. They also demonstrated behavioral defects and further noted macrocephaly. Two subsequent publications from the same group showed impairment in adipogenesis[79] and in oligodendrocyte myelination[80], causing a slender habitus and behavioral defects, respectively. In 2017, a CRISPER-mediated knockout mouse study also showed macrocephaly and widespread gene expression profile changes, enriched for neurogenesis and synaptic processing among others[66]. However, this study was not able to recapitulate the repetitive behaviors observed in other mouse models, but did show cognitive impairment[66]. Another CRISPER-mediated Chd8 heterozygous mouse study showed a similar widespread gene expression perturbation in the brain, macrocephaly, and in this case also learning deficits[68]. Macrocephaly was a key observation in another mouse model published the following year[69], but no behavioral defects were noted. Finally sex-specific behavioral traits in male mice but not female mice were reported[70], probing the known sex bias for ASD presentation.
As we noted for the human cellular model studies, there is a discrepancy in results for the mouse models. The molecular, physiological and behavioral characterizations conducted by each group were not the same, and hence, we cannot make definitive conclusions about the lack of sameness. However, it is remarkable that macrocephaly was commonly observed. Behavioral phenotypes were also reported by the majority of the groups.
We caution that the findings reported from these studies are a consequence of their setting out to study one condition only, viz, ASD, and therefore they do not rule out a wider role for Chd8 deficiency. Finally, we note that as is presumed for humans, the mouse homozygous deletion is embryonic lethal, which serves to underscore the importance of Chd8 in development.
Conclusion
The importance of correct epigenetic regulation and normal epigenomic state as integral to normal nervous development and function is now recognized[30,39,53]. Among the best known epigenomic regulatory processes, chromatin remodeling or modification is gaining significance as a key transcriptional regulatory process with widespread activity. The CHDs are a group of nine proteins encoded by the corresponding genes CHD1-9, which comprise one of the four main chromatin remodeling protein families, viz., the chromodomain helicase DNA-binding protein family. Of the nine CHDs, five are currently known to cause autosomal dominant syndromic NDDs with a diverse range of phenotypes including ID, ASD, neurological defects, skeletal defects, macrocephaly, and other major congenital anomalies. The fact that all five known CHD syndromes are autosomal dominant underscores the importance of these proteins in development.
The remarkable range of phenotypes presented, however, also indicates that the CHDs have widespread developmental roles. Though a number of cellular and model organism studies have attempted to map the molecular role of CHDs, their results also indicate a broad role for CHDs as epigenomic programmers. As we have laid out at the beginning of this review, epigenetic regulation is a complex process involving several key mechanisms that do not act alone, but rather as a concert of processes able to provide fine-tuned response to environmental states and developmental goals. It is important to keep this in mind when discussing the role of any one or a class of epigenetic regulatory proteins. Thus, given these contexts, extracting the precise pathophysiological roles of any one of the CHDs will be challenging.
In this paper, we overviewed the ability of the environment to influence the epigenome, highlighting the notion that therefore, externally induced environmental changes may hold promise to correct constitutional genetic insults that perturb the normal epigenome. However, we are as yet unaware of any results demonstrating that such a curative strategy may have an impact on NDDs caused by CHD defects. Rather, the widespread and fundamental nature of CHD control on development argues against easily being able to environmentally correct CHD genetic defects. Nevertheless, we end this review by calling for such exploratory studies in model systems. A large number of reports in the past two decades have proven that CHDs have a key regulatory function in development and control, which therefore prompts the call for creative and wide-ranging research into possible therapeutic methods.
Declarations
Authors’ contributionsProvided figure and assisted with literature review: Yasin H
Conceived and wrote the manuscript: Zahir FR
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
2. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806-11.
3. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet 2013;14:204-20.
4. Walsh CP, Xu GL. Cytosine methylation and DNA repair. Curr Top Microbiol Immunol 2006;301:283-315.
5. Medvedeva YA, Khamis AM, Kulakovskiy IV, Ba-Alawi W, Bhuyan MSI, et al. Effects of cytosine methylation on transcription factor binding sites. BMC genomics 2014;15:119.
6. Patil V, Ward RL, Hesson LB. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 2014;9:823-8.
7. Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 2013;23:555-67.
8. Shayevitch R, Askayo D, Keydar I, Ast G. The importance of DNA methylation of exons on alternative splicing. RNA 2018;24:1351-62.
9. Trivedi M, Deth R, Zhang Y, Abdolmaleky H. DNA methylation, hydroxymethylation and formylation in human frontal cortex of autistic and schizophrenic subjects. Faseb J 2017;31:468.3.
10. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011;21:381-95.
11. Bogliotti YS, Ross PJ. Mechanisms of histone H3 lysine 27 trimethylation remodeling during early mammalian development. Epigenetics 2012;7:976-81.
12. Liu X, Wang C, Liu W, Li J, Li C, et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 2016;537:558-62.
13. Miller JL, Grant PA. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell Biochem 2013;61:289-317.
14. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem 2009;78:273-304.
15. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med 2017;49:e324.
16. Micucci JA, Sperry ED, Martin DM. Chromodomain helicase DNA-binding proteins in stem cells and human developmental diseases. Stem Cells Dev 2015;24:917-26.
17. Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 2010;11:285-96.
18. Rudnizky S, Malik O, Bavly A, Pnueli L, Melamed P, et al. Nucleosome mobility and the regulation of gene expression: insights from single-molecule studies. Protein Sci 2017;26:1266-77.
19. Ehrenhofer-Murray AE. Chromatin dynamics at DNA replication, transcription and repair. Eur J Biochem 2004;271:2335-49.
20. Swygert SG, Peterson CL. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Biophys Acta - Gene Regul Mech 2014;1839:728-36.
21. Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol 2000;20:1899-910.
22. Tyagi M, Imam N, Verma K, Patel AK. Chromatin remodelers: We are the drivers!! Nucleus 2016;7:388-404.
23. Li W, Mills AA. Architects of the genome: CHD dysfunction in cancer, developmental disorders and neurological syndromes. Epigenomics 2014;6:381-95.
25. Hall JA, Georgel PT. CHD proteins: a diverse family with strong ties. Biochem Cell Biol 2007;85:463-76.
26. Delmas V, Stokes DG, Perry RP. A mammalian DNA-binding protein that contains a chromodomain and an SNF2/SWI2-like helicase domain. Proc Natl Acad Sci U S A 1993;90:2414-8.
27. Sanchez R, Zhou MM. The PHD finger: a versatile epigenome reader. Trends Biochem Sci 2011;36:364-72.
28. Xue Y, Wong J, Moreno GT, Young MK, Côté J, et al. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell 1998;2:851-61.
29. Lamar KMJ, Carvill GL. Chromatin remodeling proteins in epilepsy: lessons from CHD2-associated epilepsy. Frontiers in Molecular Neuroscience 2018;11:208.
30. Zahir FR, Brown CJ. Epigenetic impacts on neurodevelopment: pathophysiological mechanisms and genetic modes of action. Pediatr Res 2011;69:92R-100.
31. van Loo KMJ, Martens GJM. Genetic and environmental factors in complex neurodevelopmental disorders. Curr Genomics 2007;8:429-44.
32. Chelly J, Khelfaoui M, Francis F, Chérif B, Bienvenu T. Genetics and pathophysiology of mental retardation. Eur J Hum Genet 2006;14:701-13.
33. Shashi V, McConkie-Rosell A, Rosell B, Schoch K, Vellore K, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med 2014;16:176-82.
34. Tucker T, Zahir FR, Griffith M, Delaney A, Chai D, et al. Single exon-resolution targeted chromosomal microarray analysis of known and candidate intellectual disability genes. Eur J Hum Genet 2014;22:792-800.
35. Zoghbi HY, Beaudet AL. Epigenetics and human disease. Cold Spring Harb Perspect Biol 2016;8:a019497.
37. Zahir FR, Langlois S, Gall K, Eydoux P, Marra MA, et al. A novel de novo 1.1 Mb duplication of 17q21.33 associated with cognitive impairment and other anomalies. Am J Med Genet Part A 2009;149A:1257-62.
38. Kordi-Tamandani DM, Sahranavard R, Torkamanzehi A. Analysis of association between dopamine receptor genes’ methylation and their expression profile with the risk of schizophrenia. Psychiatr Genet 2013;23:183-7.
39. Bastaki KN, Alwan S, Zahir FR. Maternal prenatal exposures in pregnancy and autism spectrum disorder: an insight into the epigenetics of drugs and diet as key environmental influences. Adv Neurobiol 2020;24:143-62.
40. Buss C, Entringer S, Swanson JM, Wadhwa PD. The role of stress in brain development: the gestational environment’s long-term effects on the brain. Cerebrum 2012;2012:4.
41. Kundakovic M, Jaric I. The epigenetic link between prenatal adverse environments and neurodevelopmental disorders. Genes (Basel) 2017;8:104.
42. Monk C, Spicer J, Champagne FA. Linking prenatal maternal adversity to developmental outcomes in infants: the role of epigenetic pathways. Dev Psychopathol 2012;24:1361-76.
43. Kim DR, Bale TL, Epperson CN. Prenatal programming of mental illness: current understanding of relationship and mechanisms. Curr Psychiatry Rep 2015;17:5.
44. Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol 2011;31:363-73.
45. Breton-Larrivée M, Elder E, McGraw S. DNA methylation, environmental exposures and early embryo development. Anim Reprod 2019;16:465-74.
47. Hsieh J, Gage FH. Chromatin remodeling in neural development and plasticity. Curr Opin Cell Biol 2005;17:664-71.
48. Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem 2006;75:243-69.
49. Kleefstra T, Kramer JM, Neveling K, Willemsen MH, Koemans TS, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet 2012;91:73-82.
50. Vissers LELM, Van Ravenswaaij CMA, Admiraal R, Hurst JA, De Vries BBA, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004;36:955-7.
51. Zahir F, Firth HV, Baross A, Delaney AD, Eydoux P, et al. Novel deletions of 14q11.2 associated with developmental delay, cognitive impairment and similar minor anomalies in three children. J Med Genet 2007;44:556-61.
52. Yasin H, Gibson WT, Langlois S, Stowe RM, Tsang ES, et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J Hum Genet 2019;64:271-80.
53. Kleefstra T, Schenck A, Kramer JM, van Bokhoven H. The genetics of cognitive epigenetics. Neuropharmacology 2014;80:83-94.
54. Pilarowski GO, Vernon HJ, Applegate CD, Boukas L, Cho MT, et al. Missense variants in the chromatin remodeler CHD1 are associated with neurodevelopmental disability. J Med Genet 2018;55:561-6.
55. Snijders Blok L, Rousseau J, Twist J, Ehresmann S, Takaku M, et al. CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat Commun 2018;9:4619.
56. Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Al Turki SH, et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet 2016;48:1060-5.
57. Carvill G, Helbig I, Mefford H. CHD2-related neurodevelopmental disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. Seattle: University of Washington; 2015.
58. Martin DM. Epigenetic developmental disorders: CHARGE syndrome, a case study. Curr Genet Med Rep 2015;3:1-7.
59. Hartshorne TS, Hefner MA, Davenport SLH. Behavior in CHARGE syndrome: Introduction to the special topic. Am J Med Genet Part A 2005;133A:228-31.
60. Pagon RA, Graham JMJ, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr 1981;99:223-7.
61. Hota SK, Bruneau BG. ATP-dependent chromatin remodeling during mammalian development. Development 2016;143:2882-97.
62. Damaschke NA, Yang B, Blute ML, Lin CP, Huang W, et al. Frequent disruption of chromodomain helicase DNA-binding protein 8 (CHD8) and functionally associated chromatin regulators in prostate cancer. Neoplasia 2014;16:1018-27.
63. Sawada G, Ueo H, Matsumura T, Uchi R, Ishibashi M, et al. CHD8 is an independent prognostic indicator that regulates Wnt/β-catenin signaling and the cell cycle in gastric cancer. Oncol Rep 2013;30:1137-42.
64. Wade AA, Lim K, Catta-Preta R, Nord AS. Common CHD8 genomic targets contrast with model-specific transcriptional impacts of CHD8 haploinsufficiency. Front Mol Neurosci 2019;11:481.
65. Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, et al. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 2016;19:1477-88.
66. Gompers AL, Su-Feher L, Ellegood J, Copping NA, Riyadh MA, et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat Neurosci 2017;20:1062-73.
67. Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, et al. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016;537:675-9.
68. Platt RJ, Zhou Y, Slaymaker IM, Shetty AS, Weisbach NR, et al. Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep 2017;19:335-50.
69. Suetterlin P, Hurley S, Mohan C, Riegman KLH, Pagani M, et al. Altered neocortical gene expression, brain overgrowth and functional over-connectivity in Chd8 haploinsufficient mice. Cereb Cortex 2018;28:2192-206.
70. Jung H, Park H, Choi Y, Kang H, Lee E, et al. Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat Neurosci 2018;21:1218-28.
71. Wong WR, Brugman KI, Maher S, Oh JY, Howe K, et al. Autism-associated missense genetic variants impact locomotion and neurodevelopment in Caenorhabditis elegans. Hum Mol Genet 2019;28:2271-81.
72. Gervais L, van den Beek M, Josserand M, Sallé J, Stefanutti M, et al. Stem cell proliferation is kept in check by the chromatin regulators Kismet/CHD7/CHD8 and Trr/MLL3/4. Dev Cell 2019;49:556-73.e6.
73. Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014;158:263-76.
74. Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci 2014;111:E4468-77.
75. Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism 2017;8:1-17.
76. Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism 2015;8:1-18.
77. Wilkinson B, Grepo N, Thompson BL, Kim J, Wang K, et al. The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl Psychiatry 2015;5.
78. Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 2015;162:375-90.
79. Kita Y, Katayama Y, Shiraishi T, Oka T, Sato T, et al. The autism-related protein CHD8 cooperates with C/EBPβ to regulate adipogenesis. Cell Rep 2018;23:1988-2000.
80. Kawamura A, Katayama Y, Nishiyama M, Shoji H, Tokuoka K, et al. Oligodendrocyte dysfunction due to Chd8 mutation gives rise to behavioral deficits in mice. Hum Mol Genet 2020;29:1274-91.
81. Weiss K, Terhal PA, Cohen L, Bruccoleri M, Irving M, et al. De Novo Mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am J Hum Genet 2016;99:934-41.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yasin H, Zahir FR. Chromodomain helicase DNA-binding proteins and neurodevelopmental disorders. J Transl Genet Genom 2020;4:307-19. http://dx.doi.org/10.20517/jtgg.2020.30
AMA Style
Yasin H, Zahir FR. Chromodomain helicase DNA-binding proteins and neurodevelopmental disorders. Journal of Translational Genetics and Genomics. 2020; 4(4): 307-19. http://dx.doi.org/10.20517/jtgg.2020.30
Chicago/Turabian Style
Yasin, Heba, Farah R. Zahir. 2020. "Chromodomain helicase DNA-binding proteins and neurodevelopmental disorders" Journal of Translational Genetics and Genomics. 4, no.4: 307-19. http://dx.doi.org/10.20517/jtgg.2020.30
ACS Style
Yasin, H.; Zahir FR. Chromodomain helicase DNA-binding proteins and neurodevelopmental disorders. J. Transl. Genet. Genom. 2020, 4, 307-19. http://dx.doi.org/10.20517/jtgg.2020.30
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 31 clicks
Cite This Article 31 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.