
Idiopathic generalized epilepsy and café-au-lait macules as the predominant features in NF1 mild form
 0
0Abstract
Neurofibromatosis type 1 (NF1) is a complex autosomal dominant neurocutaneous disorder with a variable phenotype involving multiple body systems. It is due to a mutation in the NF1 gene, which results in the production of abnormal neurofibromin protein. According to the National Institutes of Health diagnostic criteria, hyperpigmented skin markings or café-au-lait macules (CALMs), axillary freckling, Lisch nodules, and neurofibromas are characteristic NF1 features. A milder phenotype, apparently manifesting with only pigmentary skin changes, has recently been associated with the c.2970_2972 in-frame deletion. Although neurological findings, including epilepsy and neurocognitive deficits, have been frequently described as a part of the classic NF1 form, they have not been properly characterized in this milder variant. We report for the first time the case of a patient harboring the c.2970_2972del of the NF1 gene and presenting with CALMs, idiopathic generalized epilepsy, and transient brain MRI alterations (so-called “unidentified bright objects”).
Keywords
INTRODUCTION
Neurofibromatosis type 1 (NF1) (MIM No. 162200) is a complex autosomal dominant disorder involving multiple body systems (with an estimated prevalence of 1/2500-1/3000 individuals). Neurofibromin protein, encoded by NF1 gene, is highly expressed in neurons, glia, Schwann cells, and the early stage of melanocyte development[1,2]. Neurofibromin inhibits the activity of the proto-oncogene Ras, catalyzing the guanosine triphosphate (GTP)-bound Ras hydrolysis to guanosine diphosphate (GDP)-bound Ras and thus preventing tumor growth[3]. In NF1, the absence of neurofibromin leads to unopposed GTPase activity, releasing the downstream signals involved in cell proliferation and differentiation.
According to the National Institutes of Health (NIH) diagnostic criteria[4], NF1 clinical diagnosis is based on the presence of at least two typical features or only one symptom with an affected first-degree relative. Characteristic NF1 findings are hyperpigmented skin markings or café-au-lait macules (CALMs), which typically develop in the first 2 years of life, axillary or inguinal freckling, distinctive bone anomalies, Lisch nodules, and neurofibromas. Neurologic features, in particular mild neurocognitive deficits and epilepsy, have been also frequently described in NF1[5,6]. A remarkable phenotype lacking both cutaneous neurofibromas and visible plexiform neurofibromas has been associated with the in-frame c.2970_2972del[7-9] and the missense mutation at codon Arg 1809[10] mutations.
Here, we describe, for the first time, an interesting milder variant manifesting with idiopathic generalized epilepsy (IGE) and typical pigmentary skin manifestations.
CASE REPORT
We report the case of a 27-year-old, right-handed man, who first presented typical absence seizures at the age of 4. At that time, the electroencephalogram (EEG) showed 3-Hz generalized spike-and-wave discharges. At 16 years, he was hospitalized following the occurrence of a generalized tonic-clonic seizure. During his hospital stay, the EEG confirmed the previous findings and a brain magnetic resonance imaging (MRI) showed focal bilateral subcortical signal alterations in the T2-weighted sequences [Figure 1A], which had completely disappeared by the time the patient undertook a follow-up MRI scan 4 months later [Figure 1B]. In addition, during hospitalization the physicians also observed pigmentary manifestations similar to CALMs - eight café-au-lait macules of 10-20 mm located on the arms and the trunk - although no other feature suggestive of NF1 was detected.
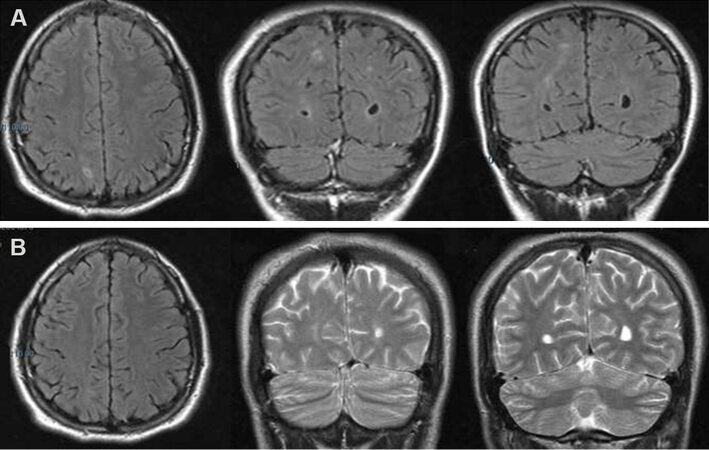
Figure 1. Brain MRI scan showing, in T2 or FLAIR-weighted images, bilateral focal subcortical alterations mostly involving the right hemisphere (A). The alterations had disappeared at MRI follow-up performed four months later (B).
At the age of 26, the patient came to our observation for seizure persistence. He underwent a video-EEG recording that showed the presence of generalized epileptiform abnormalities [Figure 2]. Brain MRI and neurologic examination were normal. A complete neuropsychological evaluation (including Phonemic/Semantic Alternate Fluency Test, Attentive Matrix, Trail Making Test, Rey Auditory Verbal Learning Test, Rey-Osterrieth Complex Figure Test, Short-Story Recall Test, Stroop Test, Raven Colored Progressive Matrices, Abstract Verbal Judgment, WAIS-IV Working Memory Scale, Tower of London Test, and Ideomotor Apraxia Test) documented a normal cognitive profile except for a slight attention shifting deficit. Seizure control was achieved thanks to a polytherapy with valproate 1500 mg/day, zonisamide
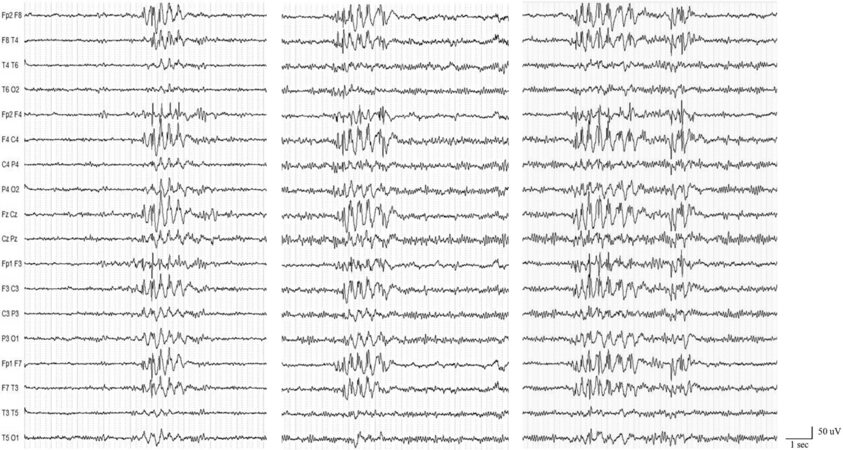
Figure 2. Video-electroencephalogram (EEG) recording showing interictal bilateral synchronous spike- and polyspike-waves discharges. A Micromed, System Plus, 21-channel device was used. EEG electrodes were placed on the scalp according to the 10-20 International System, and all tracings were interpreted using a bipolar longitudinal montage.
DISCUSSION
Our case presents a peculiar mild phenotype related to a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del) with CALMs as the sole extra-neurological sign, in line with some of previous reports[7-9,11]. In our patient, epilepsy was the main neurological manifestation while the cognitive profile was normal, contrarily to the only other case described in the literature. In that case, the c.2970_2972del was reported in a patient with psychomotor delay, learning difficulties, dyslexia, and drug-resistant epilepsy with different seizure types, suggesting a possible epileptic encephalopathy[8]. Epilepsy is commonly associated with the classic NF1 form, in which the risk of developing seizures is higher than the general population, with a prevalence of 4%-7%[6]. Seizures may be the first symptom in NF1 patients, and, in most cases, they have a focal onset. The underlying etiology may often lie in intracranial NF1-related tumors or other brain structural abnormalities, including hippocampal sclerosis and polymicrogyria[6,12,13]. Our case is peculiar first because epilepsy is occasionally described in the NF1 milder variant. Moreover, the electro-clinical features strongly support the diagnosis of IGE. This type of epilepsy was described only in few cases of classic NF1, but it has never been reported in its milder form. Because presence/absence of epilepsy and “unidentified bright objects” (UBOs) was not addressed in three of the four phenotype-genotype reports of “milder” NF1[7,9,10], it cannot be assumed that these features were absent in the subjects. Although pathophysiological mechanisms have not been clarified, it is widely acknowledged that neurofibromin is abundantly expressed in cerebral cortex during embryogenesis; therefore, a variant in NF1 gene could result in an aberrant neural network, determining an epileptic activity.
Another interesting finding observed in our case was the detection of the so-called UBOs, transient T2 hyperintensities revealed by brain MRI at disease onset. According to published data, these structural alterations, observed in up to 93% of NF1 children[14], could remain stable or disappear over several years, rarely evolving into brain gliomas[12,15,16]. The potential role of UBOs in precipitating seizures and causing epilepsy is rather controversial[12], but they are usually not considered as a risk factor for epilepsy since they almost exclusively involve white matter and basal ganglia and rarely extend to the cortex. Although it is not possible to definitely rule out a contribution of UBO in facilitating or promoting epileptogenesis and ictogenesis, our patient’s typical IGE electro-clinical presentation makes the causal relationship between UBO and epilepsy quite unlikely.
In conclusion, this case represents a further contribution to better define the neurological findings in patients with 3-bp in-frame deletion of the NF1 gene (c.2970_2972del). It appears peculiar for several reasons: (1) the neurologic phenotype included epilepsy without other neuropsychiatric deficits; and (2) the electro-clinical features were strongly suggestive of IGE (firstly described here).
Finally, our case demonstrated that the co-occurrence of epilepsy and cutaneous alterations (mainly CALMs) should prompt physicians to consider NF1 also in cases with subtle clinical manifestations. A causal relationship between IGE and NF1 c.2970_2972del cannot be ruled out, but it is also possible that the patient has IGE susceptibility variant(s) at other gene(s)[17] that could be contributing.
DECLARATIONS
Authors’ contributionsManuscript draft, data aquisition and analysis: Fanella M, Mastromoro G
Manuscript draft and revision: Morano A
Data aquisition and analysis: Tessari G, Cerulli Irelli E
Study conception, critical reappraisal: Pizzuti A, Giallonardo AT
Manuscript revision and critical reappraisal: Ferracuti S
Study conception, manuscript revision and critical reappraisal: Di Bonaventura C
Availability of data and materialsClinical data and reports supporting the results reported in this article can be found in the patient’s medical records.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationA written informed consent for publication was obtained.
Copyright© The Author(s) 2021.
REFERENCES
1. Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron 1992;8:415-28.
2. Malhotra R, Ratner N. Localization of neurofibromin to keratinocytes and melanocytes in developing rat and human skin. J Invest Dermatol 1994;102:812-8.
3. Martin GA, Viskoohil D, Bollag G, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990;63:843-9.
4. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988;45:575-8.
5. Nix JS, Blakeley J, Rodriguez FJ. An update on the central nervous system manifestations of neurofibromatosis type 1. Acta Neuropathol 2020;139:625-41.
6. Pecoraro A, Arehart E, Gallentine W, et al. Epilepsy in neurofibromatosis type 1. Epilepsy Behav 2017;73:137-41.
7. Koczkowska M, Callens T, Gomes A, et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): an update of genotype-phenotype correlation. Genet Med 2019;21:867-76.
8. Quintáns B, Pardo J, Campos B, et al. Neurofibromatosis without neurofibromas: confirmation of a genotype-phenotype correlation and implications for genetic testing. Case Rep Neurol 2011;3:86-90.
9. Upadhyaya M, Huson SM, Davies M, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet 2007;80:140-51.
10. Rojnueangnit K, Xie J, Gomes A, et al. High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum Mutat 2015;36:1052-63.
11. Muram-Zborovski TM, Vaughn CP, Viskochil DH, Hanson H, Mao R, Stevenson DA. NF1 exon 22 analysis of individuals with the clinical diagnosis of neurofibromatosis type 1. Am J Med Genet A 2010;152A:1973-8.
12. Hsieh HY, Fung HC, Wang CJ, Chin SC, Wu T. Epileptic seizures in neurofibromatosis type 1 are related to intracranial tumors but not to neurofibromatosis bright objects. Seizure 2011;20:606-11.
13. Kuroda N, Fujimoto A, Okanishi T, Sato K, Nishimura M, Enoki H. Epilepsy surgery for a patient with neurofibromatosis type 1 concomitant with moyamoya syndrome. J Clin Neurosci 2019;61:307-10.
14. Griffiths PD, Blaser S, Mukonoweshuro W, Armstrong D, Milo-Mason G, Cheung S. Neurofibromatosis bright objects in children with neurofibromatosis type 1: a proliferative potential? Pediatrics 1999;104:e49.
15. Griffith JL, Morris SM, Mahdi J, Goyal MS, Hershey T, Gutmann DH. Increased prevalence of brain tumors classified as T2 hyperintensities in neurofibromatosis 1. Neurol Clin Pract 2018;8:283-91.
16. Billiet T, Mädler B, D'Arco F, et al. Characterizing the microstructural basis of “unidentified bright objects" in neurofibromatosis type 1: A combined in vivo multicomponent T2 relaxation and multi-shell diffusion MRI analysis. Neuroimage Clin 2014;4:649-58.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Fanella M, Mastromoro G, Morano A, Cerulli Irelli E, Tessari G, Ferracuti S, Giallonardo AT, Pizzuti A, Di Bonaventura C. Idiopathic generalized epilepsy and café-au-lait macules as the predominant features in NF1 mild form. J Transl Genet Genom 2021;5:124-9. http://dx.doi.org/10.20517/jtgg.2021.11
AMA Style
Fanella M, Mastromoro G, Morano A, Cerulli Irelli E, Tessari G, Ferracuti S, Giallonardo AT, Pizzuti A, Di Bonaventura C. Idiopathic generalized epilepsy and café-au-lait macules as the predominant features in NF1 mild form. Journal of Translational Genetics and Genomics. 2021; 5(2): 124-9. http://dx.doi.org/10.20517/jtgg.2021.11
Chicago/Turabian Style
Fanella, Martina, Gioia Mastromoro, Alessandra Morano, Emanuele Cerulli Irelli, Gianmarco Tessari, Stefano Ferracuti, Anna T. Giallonardo, Antonio Pizzuti, Carlo Di Bonaventura. 2021. "Idiopathic generalized epilepsy and café-au-lait macules as the predominant features in NF1 mild form" Journal of Translational Genetics and Genomics. 5, no.2: 124-9. http://dx.doi.org/10.20517/jtgg.2021.11
ACS Style
Fanella, M.; Mastromoro G.; Morano A.; Cerulli Irelli E.; Tessari G.; Ferracuti S.; Giallonardo AT.; Pizzuti A.; Di Bonaventura C. Idiopathic generalized epilepsy and café-au-lait macules as the predominant features in NF1 mild form. J. Transl. Genet. Genom. 2021, 5, 124-9. http://dx.doi.org/10.20517/jtgg.2021.11
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.