
The (epi)genomic landscape of splenic marginal zone lymphoma, biological implications, clinical utility, and future questions

Abstract
Splenic marginal zone lymphoma (SMZL) is an indolent B-cell lymphoma comprising less than 2% of lymphoid neoplasms. Approximately 70% of patients have a progressive disease requiring treatment and up to 30% of patients relapse or transform to diffuse large B-cell lymphoma. Whilst research over the last decade has transformed our understanding of many B-cell tumours, it is only beginning to shed light on the molecular pathogenesis of SMZL. Expansive immunogenetic investigations have shown biases in the immunoglobulin gene repertoire with distinct patterns of somatic hypermutation, suggesting a pathogenic role for antigen selection. In parallel cytogenetic studies have found a number of recurrent chromosomal lesions, in particular a deletion of the long arm of chromosome 7, though causative genes have not been identified. Our understanding of the mutational landscape of SMZL is built on a limited number of index cases, but has highlighted recurrent mutations in KLF2, NOTCH2 and TP53, and a spectrum of genes that cluster within biological pathways of importance in B-cell differentiation. While preliminary DNA methylation profiling has shown epigenetically distinct patient sub-groups, including a group defined by elevated expression of polycomb repressor complex 2 components. This review will provide an overview of our current understanding of the molecular basis of SMZL, and how this information impacts patient outcomes. Furthermore, we will outline; (1) the knowledge gaps that still exist; (2) a potential future research direction; and (3) how a detailed molecular understanding of the disease will ultimately provide patients with improved management and treatment choices.
Keywords
INTRODUCTION
The World Health Organization classification of tumours of the hematopoietic and lymphoid tissues defines three marginal zone (MZ) lymphoma entities, splenic marginal zone lymphoma (SMZL), nodal MZL (NMZL) and extranodal MZL[1]. In addition, a number of provisional entities are emerging; these include splenic diffuse red pulp lymphoma (SDRPL), hairy cell leukaemia-variant (HCL-v) and clonal B-cell lymphocytosis of marginal zone origin (CBL-MZ), the latter of which is clonally related to SMZL in a proportion of cases[2-5]. SMZL comprises less than 2% of lymphoid neoplasms, typically involving the spleen, bone marrow and peripheral blood, although a minority of patients have low volume extra hilar intra-abdominal lymphadenopathy detectable on CT scanning. Patients most commonly present with abdominal discomfort due to massive splenomegaly, anaemia secondary to hypersplenism or auto-immune haemolysis, or incidentally due to either clinical or radiological detection of asymptomatic splenomegaly or an abnormal blood count. The median age of diagnosis is 65 and patients exhibit a 10-year median survival time[6,7]. The diagnosis is most secure in patients who undergo splenectomy, now performed almost always for therapeutic reasons, but it increasingly relies on a combination of clinical features together with an assessment of lymphocyte morphology and immunophenotype, bone marrow histology and immunohistochemistry[7]. In the absence of splenic histology, the differential diagnosis from common low-grade B-cell disorders such as chronic lymphocytic leukaemia (CLL), follicular lymphoma (FL), and mantle cell lymphoma (MCL) is usually straightforward but SMZL lacks a disease-specific immunophenotype and the distinction between SMZL and some cases of HCL-v, SDRPL and lymphoplasmacytic lymphoma (LPL) may be more difficult. Approximately 70% of patients have a progressive disease requiring treatment. Standard treatment options include splenectomy, Rituximab or Rituximab and bendamustine, all of which achieve high response rates with prolonged progression-free survival[8,9]. However, up to 30% of patients relapse and are intolerant of, or resistant to, standard therapies or transform to diffuse large B-cell lymphoma (DLBCL) with dismal survival[10]. Therefore, there is an unmet need for well tolerated, preferably oral, novel therapies.
Whilst SMZL is still a relatively under-studied malignancy, several works published over the last decade or so have begun to unravel the intrinsic molecular defects present in the SMZL B-cell, and extrinsic cellular mechanisms that reflect micro-environmental and antigenic interactions [Figure 1]. This review will focus on providing a summary of the (epi)genetic and immunogenetic landscape of the disease, the associated clinico-biological implications of these aberrations, as well as supplying an overview of key unanswered questions that might direct future research in this area.
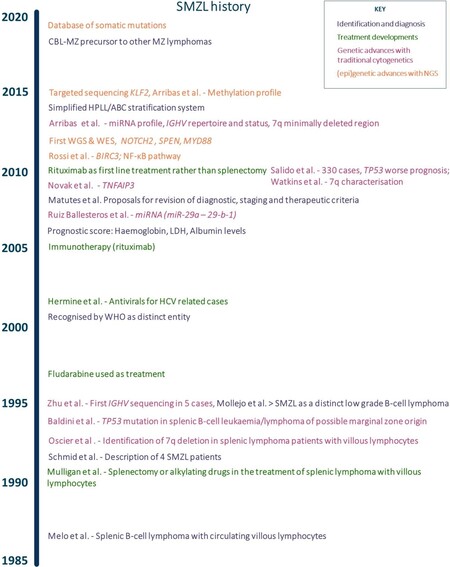
Figure 1. Timeline highlighting historical milestones in the understanding of SMZL. HPLL refers to the determinant factors in the stratification model (haemoglobin, platelet count, high lactate dehydrogenase, and lymphadenopathy) and the three groups patients are grouped in (A, B and C). NGS: Next generation sequencing; CBL-MZ: clonal B-cell lymphocytosis of marginal zone origin; MZ: marginal zone; IGHV: immunoglobulin heavy chain variable region; WGS: whole genome sequencing; WES: whole exome sequencing; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; LDH: lactate dehydrogenase; HCV: hepatitis C virus; WHO: World Health Organisation; SMZL: splenic marginal zone lymphoma. Additional references[11-19].
ANALYSIS OF THE IMMUNOGLOBULIN GENES
The most important molecule expressed on the cell surface of SMZL B-cells is the B-cell receptor (BCR), which allows interaction with antigens and antigenic elements within the tumour microenvironment. Seminal sequencing studies of the immunoglobulin (IG) have been performed on the myriad of mature B-cell neoplasms, including SMZL, where they have identified key features of the B-cell receptor immunoglobulin repertoire indicating that clonal B-cell selection by antigen/superantigens is an important feature of SMZL pathophysiology. Investigation of large SMZL cohorts has demonstrated remarkably biased usage of immunoglobulin heavy chain (IGHV) genes, namely enrichment of the IGHV1-02 (30%), IGHV4-34 (11%) and IGHV3-23 (9%) genes, which collectively represent almost half of SMZL cases[20,21]. What is noteworthy is the scarcity of the IGHV1-02 gene in other mature B-cell tumours, even tumours of the spleen, such as SDRPL[20], suggesting that a distinctive selection process may predominate in SMZL pathogenesis.
The majority (90%) of IGHV1-02 patients are IGHV1-02*04[20-22] with extended restriction to the level of polymorphic variation. Evidence of somatic hypermutation (SHM) is seen in the majority of IGHV1-02*04 SMZL cases (~95%) suggesting exposure to antigen is both important in progenitor tumour cell selection but also relevant to ongoing evolution[22]. IGHV1-02*04 cases carry extended VH CDR3 sequences, biased IGHD and IGHJ gene usage, and distinct patterns of SHM[20]. IGHV1-02 cases with allele *04 may have unique immunogenetic features as the polymorphism encodes a tryptophan (W) residue rather than arginine (R) at position 75 of VH FR3, with the latter playing a role in IG structural stability and conformation. As will be discussed in more detail later, key somatic mutations and acquired copy number changes define the disease but are also associated with specific immunological backgrounds. What is the most notable is the enrichment of 7q deletions and KLF2 and NOTCH2 mutations in IGHV1-02*04 SMZL[23,24]. These analyses, with preliminary DNA methylation and transcriptomic studies[25,26], suggest cases with IGHV1-02*04 represent a distinct patient sub-group, which is likely to have emerged from a cell with a distinct ancestry and/or unique immune activation process followed by transformation, ongoing antigen exposure, with shared genomic lesions and poor survival[24].
The critical part that the BCR plays in SMZL is further emphasized by the presence of somatic hypermutations, as only 12% of cases lack any evidence of SHM at the IGHV locus, and might be considered truly unmutated[20,21]. The remaining cases exhibit evidence of SHM at the IGHV locus, with 38% and 50% defined as borderline (97%-99.9% IGHV gene identity to germline) and significantly mutated (< 97% IGHV gene identity to germline), respectively. The majority (71%) of IGHV1-02*04 cases harbour borderline levels of SHM, often benign in nature that cluster in the framework region[20,22]. Whilst levels of SHM represent a critically important prognostic and predictive biomarker in CLL[27,28], their clinical utility in SMZL is less established. Using SHM cut-offs established for CLL, although SHM levels are likely to be disease-specific, showed no difference in progression-free and overall survival between mutated and unmutated IGHV cases[29,30]. In contrast, we have shown that a physiological definition, based on the complete absence of SHM (truly unmutated), is able to provide independent prognostic information, with truly unmutated cases exhibiting reduced time to treatment[24]. Taken together, these observations show that we continue to lack a definitive understanding of the clinical utility of SHM in SMZL, likely the result of small patient cohorts, heterogeneous treatment, and an inaccurate definition of the most biologically and clinically relevant SHM cut-offs.
SOMATIC GENOMIC ABERRATIONS
The largest reported study of cytogenetic abnormalities in SMZL comprised 330 untreated cases of whom 143 had undergone splenectomy[31]. 72% displayed an abnormal karyotype using conventional chromosomal banding, with 50% exhibiting a complex karyotype (defined as three or more cytogenetic aberrations). The majority of aberrations resulted in copy number abnormalities (CNAs) with chromosomal gains exceeding losses. The most frequent gains were: 3/3q, trisomy 18 and trisomy 12 comprising 25%, 12% and 8% of abnormal karyotypes, respectively. The commonest deletions involved 7q, 6q and 13q in 39%, 11.7% and 5% of cases respectively [Table 1]. 18% of cases had concomitant deletion of 7q and gain of 3q in the same karyotype. Interestingly, the use of FISH probes and single nucleotide polymorphism (SNP) arrays has shown that the loci of genes recurrently mutated in SMZL, such as KLF2, BIRC3 and TRAP3 may also be the site of small mono-allelic deletions[32,33]. In contrast, the potential functional consequences of trisomies, whole arm gains or large deletions are much more difficult to elucidate. Generally, chromosomes that are repeatedly gained are enriched for genes that promote cell proliferation and similar aneuploidy patterns are seen in tumours derived from the same cell types[34,35]. The latter observation would be consistent with the frequency of trisomy 3, 12 and 18 across low grade B cell tumours.
Summary of recurrent chromosomal aberrations in SMZL
| Chromosome | Frequency | Target genes/clinico-biological associations |
| 7q- | 14%-44% | Unknown target gene(s) High frequency in SMZL compared to other MZL[29,36] Associated with unmutated IGHV genes, IGHV1-2*04 usage, and KLF2 and NOTCH2 somatic mutations[24,31,37] |
| 17p- | 5%-32% | TP53 gene Associated with worse prognosis in univariate analysis[31] |
| 6q- | 8%-24% | A fraction of cases include deletion of TNFAIP3 (negative regulator of NF-κB) |
| 8p- | 4%-15% | Associated with poor outcome in MZLs[29] No link with outcome unless co-existing with deletion of 17p[29] |
| 13q- | 5%-18% | SMZL cases with this lesion showed a genetic profile consistent with SMZL diagnosis[29] |
| 14q- | 3%-10% | Linked to inferior prognosis but detected in the context of a complex karyotype[31] |
| +3/3q+ | 15%-34% | Two gained regions, one included gene BCL6 located at 3q27 Associated with complex karyotypes Tend to occur in cases without del 7q 3q gains have been associated with gains at 1q and 17q22-q25.3, del 6q23.2-q24.1 (TNFAIP3) and del 6q25[29] |
| +12/12q+ | 8%-25% | Trisomy 12 & use of heavy chain VH3 family gene segment were found significantly associated with worse OS in univariate analysis but lost significance in multivariate analysis[36] Associated with gain of chromosome 3[31] |
| +18/18q+ | 8%-23% | Mutually exclusive to 7q deletions[36] Associated with gain of chromosome 3[31] |
| 8q+ | 2%-20% | Gains of 8q that include MYC gene locus were associated with poor clinical outcomes[38] |
Studies of 5q deletions in myelodysplastic syndromes and acute myeloid leukaemia illustrate both the heterogeneity of their genomic landscape and provide a paradigm for the investigation of large deletions. However, unlike with 7q deleted in SMZL patients, different 5q minimally deletion regions (MDR) are associated with distinct clinical phenotypes which aided in the identification of causative genes[39,40]. Deletions of 7q are rare in chronic lymphoid malignancies apart from SMZL and HCL-v. In SMZL, there is no 7q minus syndrome comparable to 5q minus and 7q deletions have no obvious prognostic significance. However, 7q deletion is associated with a number of other biological features such as IGHV1-02*04 usage, mutations of NOTCH2 and KLF2 and a specific DNA methylation signature that will be outlined later in this review. Intriguingly, splenic, and nodal marginal lymphomas share remarkably similar genomic landscapes other than the presence of 7q deletions in the former and PTPRD mutations in the latter. Numerous studies have shown that deletion breakpoints at 7q are heterogeneous, with q21 the most proximal and q36 the most terminal breakpoints[19,31,41-45]. Using array comparative genomic hybridization (aCGH) in patient samples or cell lines, 2 studies identified remarkably similar MDR of 2.7-2.8 Mb in 7q32.1-q32.2[36,38]. Compared to SMZL cases without deletion of 7q, expression of coding genes and micro-RNAs (miRNAs) within the MDR’s is reduced but neither study identified microdeletions nor pathogenic mutations or disease-specific promotor hypermethylation of genes in the retained allele that would indicate a classical tumour suppressor gene. A further aCGH study showed another 1.51 Mb MDR at q22.1 in 49% of cases studied[46] which included CUX1, a gene implicated in tumorigenesis. While it is possible that 7q deletions have no impact on phenotype it seems more likely that future studies integrating several omic technologies as well as a genome wide approach will be necessary to fully comprehend the role of the 7q deletion in SMZL pathogenesis.
Recurring translocations are much less frequent than in other low-grade lymphomas such as MCL, FL and mucosa associated lymphoid tissue lymphoma, and usually involve the IGHV locus. These include t(14;19)(q32;q13), t(6;14)(p21;q32), t(9;14)(p13;q32), t(1;14)(q21;q32) which target the genes BCL3, CCND3, PAX5 and BCL9/MUC1, respectively. Translocations of 7q21, have been documented in some cases, predominantly with the immunoglobulin kappa locus resulting in upregulation of CDK6[47].
MUTATIONS IN KEY GENES AND PATHWAYS
Whole genome/exome sequencing (WGS/WES) has been employed to scan the entire cancer genome at base-pair resolution and identify somatically acquired gene mutations across a broad spectrum of mature B-cell tumours[48-52]. The analysis of large patient cohorts, often conducted as part of large international sequencing consortia, has provided a wealth of genomic information on these neoplasms, identifying a plethora of germ-line SNPs associated with disease-risk, panels of genes targeted by somatic mutations and a number of acquired mutational mechanisms. Unlike more prevalent mature B-cell malignancies, SMZL is currently precluded from these international sequencing projects, the result being an incomplete catalogue of tumour associated genomic lesions and mutational processes, drawn from a paucity of published genomic data. Only six genome-wide studies have been conducted on only 35 patients[53]. The only study employing WGS was performed by Kiel et al.[54] but was limited to six cases without matched germline DNA. Five WES studies have been carried out on discovery cases, often with subsequent targeted re-sequencing of relevant genes in additional samples. To date, none of these studies have reported mutational signatures nor mechanisms, such as kataegis and chromothripsis[55]. Even the somatic mutational burden remains disputed, with a range of somatic mutations per patients of between 9 and 82 (mean 25)[23,56,57]. This limited agreement is likely due to the low patient numbers, and experimental and computational differences in WGS/WES processing, but could also allude to disease heterogeneity and statistical power insufficient to catalogue the complete mutational landscape of the disease[23,53]. Targeted re-sequencing approaches have helped elucidate recurrently mutated genes, but these studies have often included only small numbers of matched germ-line material for analysis. Whilst we currently have only a limited picture of the somatic landscape of SMZL, several recurrently mutated genes have been identified, which preferentially target physiologically important cellular processes, such as MZ B-cell maturation and migration, and cell cycle control [Figure 2]. A number of genomic studies, with limited functional validation, all agree that the most important recurrently mutated genes are KLF2, NOTCH2 and TP53.
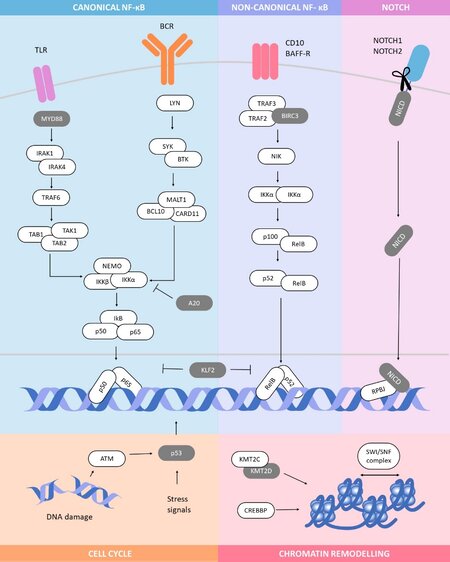
Figure 2. Main pathways targeted by somatic mutations in SMZL. Recurrently mutated genes in SMZL preferentially target physiologically important pathways, including the Notch pathway, chromatin remodelling, cell cycle control and canonical and non-canonical NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) activation. NF-κB can be activated through BCR, TLR and BAFF-R signalling. Genes encoding proteins in grey have a mutational frequency greater than 10% within SMZL cohorts. SMZL: Splenic marginal zone lymphoma; BCR: B-cell receptor; TLR: toll-like receptor; BAFF-R: B-cell activating factor receptor.
KLF2 mutations are present in up to 40% of SMZL cases
KLF2 is the most frequently mutated gene in SMZL (20%-40% of cases)[24,33,58,59]. The gene belongs to the family of Kruppel-like transcription factors, a subfamily of the zinc-finger class of DNA binding transcriptional regulators[60]. KLF2 directly binds to promoters regulating the expression of genes involved in cell homing, NF-κB signalling and cell cycle control[23,61]. In murine systems, loss of KLF2 drives the germinal cells to a MZ-like phenotype and preclusion of migration to the splenic MZ[62-64], thereby preventing germinal centre B-cell responses to antigens in the MZ. Nuclear localization of the KLF2 protein and consequent DNA binding require three C-terminal highly conserved zinc finger domains and two nuclear localization sequences, respectively. Gene mutations can be missense substitutions or truncating events [Figure 3], where the latter often results in the removal of the nuclear localization sequences[23,24,33,58]. Missense substitutions result in amino acid changes within the nuclear localization sequences of KLF2 or within highly conserved regions of the first zinc finger domain. These KLF2 mutants render the protein unable to elicit its transcriptional activity by displacement from the nucleus thereby preventing the ability of KLF2 to supress NF-κB induction by upstream signalling pathways[23,33]. The p.Q24X (stopgain) variant is a hotspot mutation[23,24,33,59]. Although no functional evidence on this specific variant is available, it is very likely that due to its position on the first exon it would result in a truncated and non-functional protein. Furthermore, this variant has a scaled Combined Annotation Dependent depletion[65] score of 36 indicating that it is predicted to be the 0.1% most deleterious substitutions you can do to the human genome. KLF2 mutations are early, clonal events, enriched in patients with deletions of 7q and IGHV1-2*04 gene usage and are associated with a short median time to first treatment in univariate survival analysis[24].
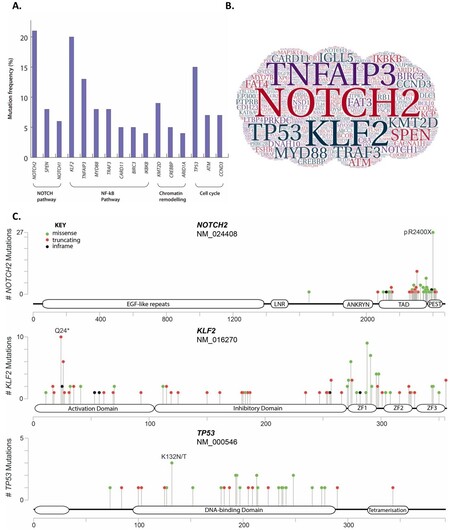
Figure 3. (A) Mutation frequency (%) of recurrently mutated genes in splenic marginal zone lymphoma (SMZL). The frequency was calculated using a systematically collated database of published mutations and genes are grouped according to affected pathways. Detailed list and description of mutations, including datasets used, can be accessed in the supplementary material from previous published work[53]. (B) Word cloud of gene symbols that have been found mutated in SMZL. The size of each gene symbol is proportional to the frequency of mutations in SMZL. (C) Lolliplot of NOTCH2, KLF2 and TP53. The figure illustrates a linear protein representing each gene with its respective domains. The height represents the number of variants reported in the literature and circle colour identifies the type of mutation. Data obtained from Jaramillo Oquendo et al.[53], 2019.
NOTCH2 mutations occur in 10%-25% of cases
In murine models, and to a lesser extent in humans also, NOTCH2 plays a key role in MZ B-cell maturation and MZ retention[66-69]. NOTCH2 is a cell-surface receptor belonging to a family of evolutionarily conserved trans-membrane proteins. Notch pathways regulate cell proliferation, cell fate, differentiation, and cell death[70]. When a ligand binds to the extracellular domain of a Notch receptor, it initiates a cascade of proteolytic cleavages that lead to the detachment of the notch intracellular domain, which then moves into the nucleus to interact with target transcription factors[70,71]. NOTCH2 is the second most frequently mutated gene present in 10%-25% of SMZL cases[23,24,54,56,72]. In SMZL, NOTCH2 mutations target the C-terminal PEST domain on exon 34[53], necessary for the regulation of the intracellular domain and consequent transcriptional regulation [Figure 3]. Shanmugam et al.[73] propose that in the case of SMZL, NOTCH2 mutations are not initiating events, but that the sustained signalling provided by these mutations provides a selective advantage in tumours that have already established themselves in a ligand-rich microenvironment. This is supported by evidence in mice where those with activating Notch2 mutations in mature B-cells displayed expansion of the marginal zone but did not develop lymphoma[74]. Additionally, NOTCH2 mutated tumours exhibit sub-clonal heterogeneity, with consequent aggressive clones[73], and inferior survival that has been demonstrated in clinical studies[24,54]. Expression of the Notch intracellular domain 2 (NICD2) can be detected in SMZL cases and is a common feature of both NOTCH2 wild-type and mutated SMZLs[73], similar to prior findings with NOTCH1 in CLL[75], suggesting that Notch activation is a general feature of SMZL tumour cells. The work by Shanmugam et al.[73] showed higher frequency of NICD2+ cells in mutated versus wild-type tumours and higher in the marginal zones of the white pulp. It is yet to be determined if enhanced NICD2 expression in wild-type tumours is explained fully by mutations in other Notch regulators, such as NOTCH1 (~5%) and SPEN (~5%)[24,57], structural or copy number aberrations, or by the enrichment of NOTCH2 in the normal counterpart of SMZL.
TP53 mutations account for up to 15% of SMZL
TP53 is one of the main SMZL associated genes implicated in cell cycle control, along with CCND3 (~5%) and ATM (~4%). However, the lack of germline material in many of the studies makes it difficult to confirm whether ATM mutations are truly somatic and should be looked at with caution. TP53 is disrupted in 10%-15% of SMZL cases[24,57]. Most TP53 mutations are missense mutations within the DNA binding domain [Figure 3], attenuating or eliminating its function as tumour suppressor, since mutant proteins lose the ability to activate canonical p53 target genes. This leads to uncontrolled cell proliferation and permissive accumulation of genomic mutations that may culminate in tumour growth[76]. Mutations in TP53 are associated with poor prognosis and are more common in cases with unmutated IGHV genes[29,31,36,38,56].
Other low prevalence genes cluster into key biological pathways
Additional recurrently mutated genes are continuing to emerge from high-throughput sequencing studies, though at lower frequencies [Table 2 and Figure 3]. As previously noted, these genes often cluster within biological pathways of known importance to B-cell differentiation and in cancer more generally. Some of the most well-established genes and pathways are summarized below:
Examples of key recurrently mutated genes in SMZL
| Gene | Frequency | Target pathway/clinico-biological associations |
| KLF2 | 12%-42% | MZ B-cell homing/NF-κB signaling Associated with 7q deletions, NOTCH2 somatic mutations and IGHV1-02*02 usage Associated with reduced TTFT in univariate analysis[24] |
| NOTCH2 | 10%- 25% | NOTCH pathway/marginal zone development Worse OS in univariate analysis. Associated with 7q deletions, KLF2 somatic mutations and IGHV1-02*04 usage Independent risk factor for TTFT (multivariate analysis)[24] |
| TP53 | ~15% | Cell cycle control/genome maintenance Associated with shorter EFS in univariate analysis but is an independent risk factor for OS (multivariate analysis)[24,38] |
| TNFAIP3 | 7%-15% | Canonical NF-κB signaling/MZ differentiation Higher frequency in small number of transformed cases[24] |
| MYD88 | 5%-15% | TLR and BCR signaling/MZ differentiation Somatic mutations associated with longer OS in univariate analysis[24] Mutually exclusive to 7q deletion, KLF2 somatic mutations and IGHV1-02*04 usage[24] |
| KMT2D | 11%-15% | Chromatin remodeling - H3K4 methylation High prevalence in other lymphomas particularly FL and DLBCL |
| TRAF3 | ~8% | Non canonical NF-κB signaling/marginal zone B-cell development |
| BIRC3 | 5%-11% | Non canonical NF-κB signaling/marginal zone B-cell development |
| NOTCH1 | ~5% | Notch pathway/marginal zone B-cell development Recurrently mutated in CLL where truncation of the PEST domain causes constitutive Notch activation |
| SPEN | ~5% | Notch pathway/marginal zone B-cell development |
| CARD11 | ~5% | BCR and NF-κB signaling/Marginal zone B-cell development |
| CREBBP | ~5% | Chromatin remodeling - BCL6 and p53 acetylation (germinal centre development) |
NF-κB signalling plays an essential role in MZ B-cell development and differentiation[77]. When normal B lymphocytes respond to antigens, NF-κB signalling is activated, reprogramming cells to favour cell cycle progression, survival, cytokine secretion and inflammation[78,79]. NF-κB activation, through either the canonical or non-canonical pathway, is transient in normal cells and depends on external stimuli including ligands for the BCR and for the Toll-like receptors[78], while termination of signalling is dependent on negative feedback mechanisms including re‐accumulation of IκBα and induction of A20[79]. Both unbiased and targeted approaches have uncovered molecular lesions affecting genes belonging to the NF-κB pathway, as well as upstream pathways connected to NF-κB activation[78]. Most notably TNFAIP3 (A20), a negative regulator of NF-κB signalling, has been found mutated in ~13% of SMZL cases[23,24,29,32,56,57,80]. Parry et al.[24] also reported higher frequency of TNFAIP3 mutations in cases that subsequently transformed, along with truly unmutated IGHV genes. Other negative regulators harbouring mutations include TRAF3 (8%) and BIRC3 (5%-11%). Both TRAF3 and BIRC3 are part of the regulatory system that negatively regulates MAP3K14, a central activator of noncanonical signalling and another target of mutations in SMZL[32]. Activating mutations in positive regulators have also been found mainly in CARD11 (~5%) and IKBKB (~3%). Furthermore, mutations in this pathway seem to be mutually exclusive pointing to multiple independent molecular mechanisms targeting NF-κB signalling in SMZLs[32].
Toll-like receptor (TLR) signalling plays a key role in SMZL biology, as cellular proliferation is driven by TLR activation. MYD88, an adaptor protein essential for proper TLR signal transduction, has several structural domains including a death domain responsible for oligomerization and interactions with IRAK1-4, which together lead to activation of NF-κB. The toll/interleukin-1 receptor homology (TIR) domain, at the proteins C-terminus, is responsible for the activation of downstream signalling. MYD88 mutations have been identified in 3%-15% of SMZL and 10% of CBL-MZ as well as across a spectrum of other B-cell tumours[24] where they occur as early clonal events, mutually exclusive to other driver mutations and in the presence of mutated IGHV genes, suggesting that these mutations define a group of homogeneous disease subtypes[23,80]. MYD88 also harbours a recurrent variant (L265P) in the TIR domain, frequently seen in a number of mature B-cell tumours[81]. This missense mutation promotes cell survival by enhancing NF-κB signalling[82]. As in CLL, MYD88 mutations have also been shown to be linked to favourable clinical outcome in SMZL using univariate approaches, though its independent prognostic relevance has yet to be confirmed[24,80].
Chromatin remodelling enzymes are dynamic modulators of cell identity and regulate B-cell differentiation and proliferation through recognition of a spectrum of specific biochemical marks on histone proteins and DNA, thereby modifying chromatin accessibility to transcription machinery proteins[83]. Recurrent somatic mutations in chromatin remodelling and epigenetic modifier genes (KMT2D 11%-15%, CREBBP ~5%, ARID1A ~4%, EP300 ~2% and TBL1XR1 ~1%) are present in ~40% of SMZL cases[24,56,57,72,84]. CREBBP mutations are often clonal mutations in SMZL[24]. Whilst the functional consequences of these mutations have not been functionally validated in SMZL, there is ample evidence in other B-cell lymphomas. Mutations in KMT2D (MLL2), a histone methyltransferase that modifies lysine-4 of histone 3, contribute to lymphomagenesis[85], with an established tumour suppressor role in DLBCL and FL[86-88]. In these same tumours, mutations in CREBBP, and more rarely, EP300, two highly similar histone acetyltransferases, result in defects in acetylation-mediated ablation of BCL6 expression and activity of p53[89]. ARID1A (BAF250a) is a component of SWI/SNF (SWItch/Sucrose Non-Fermentable) family of evolutionary conserved, multi-subunit chromatin remodelling complexes, found mutated in various cancers and in SMZL[90]. SWI/SNF regulates DNA accessibility to other proteins involved in replication and repair, allowing the activation or suppression of gene transcription[83]. It will be interesting to investigate the relationship between these mutations and any epigenetic fingerprint associated with the cell of origin in SMZL.
TRANSCRIPTOMICS
At the transcriptional level, limited gene expression profiling of SMZL cases revealed the expected signatures; most notably B-cell genes, such as those within the BCR (BTK, PKCA, NFATC1) and NF-κB (CD40, REL, BIRC3, TRAF3) pathways, and genes critical to MZ development and migration (NOTCH2)[25,91,92]. Existing data do not yield a robust gene expression signature for SMZL, but rather genes expressed across aligned mature B-cell tumours. Navarro et al.[93] constructed a gene expression classifier from a panel of B-cell tumours, which was not able to identify distinct gene signatures for SMZL, LPL and SDRPL. Of note though, a recent meta-analysis of gene expression profiles from SMZL, normal splenic material and other B-cell tumours was able to define 135 genes that discriminate SMZL from other B-cell lymphomas[94]. It will be important to validate this early observation, through the inclusion of larger patient cohorts, cell sub-population from the spleen and the inclusions of CBL-MZ patients to track progression to overt SMZL.
EPIGENETICS
Epigenetic modifications that are perturbed in cancer include dysregulation of higher order chromatin structure, such as chemical modifications to histones (thereby effecting DNA positive charge, decreasing histone/DNA affinity and altering accessibility of tertiary DNA structure), DNA methylation (regulation of gene expression by the transfer of methyl groups onto the C5 position of cytosine) and changes to miRNA expression patterns. These modifications are crucial mechanisms that control gene transcription, and genome stability, and contribute to normal B-cell maturation. DNA methylation is the epigenetic mark most well-studied in both normal and malignant B-cells, which have outlined clinically relevant changes associated with the cell-of-origin and malignant progression itself[95-99]. Murine studies first demonstrated the crucial importance of epigenetic modifiers in various aspects of B-cell differentiation[100,101] and more recently, high-resolution genome-wide analysis of sorted B-cell subsets has mapped the DNA methylome during critical stages of human B-cell development[97,102,103]. These studies suggest that the B-cells are the lineage most defined by changes in DNA methylation with up to 30% of the human DNA methylome modified during cell maturation, particularly global changes affecting heterochromatin, DNA repeats and polycomb repressor regions, and specific alterations targeting lineage defining enhancers and promoters. A number of mature B-cell tumours have been analysed at the DNA methylation level, exhibiting a global reduction in methylation levels, principally the result of hypomethylation in heterochromatic sequences that correlates with the maturation state of the putative founding normal cell type. Specific DNA hypermethylation is observed in these tumours and is highly correlated with sequences known to display polycomb repression in normal cells. Unlike other mature B-cell tumours, SMZL is under-studied at the DNA methylation level, with only a single study published that employed microarray-based promoter methylation and gene expression analysis, correlated with genomic and clinical outcome data[25].
The multicentre study performed by Arribas et al.[25] profiled the methylation status of 27,000 promoter CpGs in 134 DNA samples extracted from spleen derived SMZL tumour cells of high purity using the Infinium HumanMethylation27 arrays. These data were integrated with gene expression, copy number, somatic mutational and immunogenetic data. The authors identified a patient subgroup characterised by high genome-wide promoter methylation (High-M), inferior survival, an elevated risk of histologic transformation, and a raised prevalence of NOTCH2 mutations, 7q31-32 deletion and IGHV1-02 usage. In addition to these distinct (immuno)genetic features, the High-M subgroup was defined by significant DNA methylation and transcriptional disruption of genes involved epigenetic regulation. Most importantly, the High-M subgroup exhibited hypomethylation and consequent elevated transcription of genes involved in B-cell activation and NF-κB and those encoding for the polycomb repressor complex 2 (PRC2) components, including EED, SUZ12 and EZH2. Hypomethylation of key PRC2 components was concomitant with hypermethylation and reduced expression of PRC2 target genes and those with H3K27me3 marks, supporting the hypothesis that the expression of key differentiation genes defines this High-M sub-type[25,104]. Collectively, these data support a distinct sub-group of SMZL, driven by key IGHV usage, defined by consistent epigenetic and genomic lesions, with the likely functional impact being dysregulated lineage specification and cellular proliferation signalling [Figure 4]. The same study was performed parallel in vitro experiments with the demethylating agent, decitabine, which partially rescued key tumour suppressor genes and down-regulated survival and proliferation pathways, suggesting that the High-M sub-group might be responsive to epigenetic therapies.
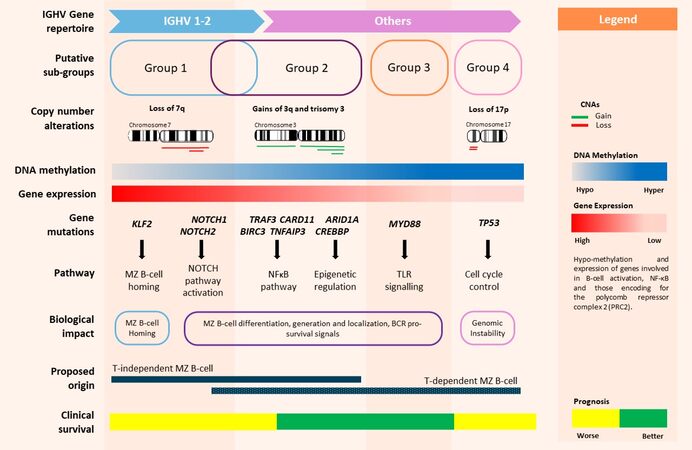
Figure 4. Putative SMZL disease subtypes. Collectively, data from different studies support a distinct sub-group of SMZL (Group 1) driven by: IGHV1-02 usage, DNA hypo-methylation and over expression of genes involved in B-cell activation, NF-κB, and those encoding for the polycomb repressor region, and recurrent genomic lesions (deletion of 7q and somatic mutations in KLF2 and NOTCH2). Sub-groups 2-4 are less well supported due to a limited number of cases and lack of functional evidence. Further work is necessary to delineate and characterise them fully. SMZL: Splenic marginal zone lymphoma; IGHV: immunoglobulin heavy chain variable region; CNAs: copy number alterations; MZ: marginal zone; TLR: toll-like receptor; BCR: B-cell receptor.
MiRNAs are a key group of short noncoding RNA molecules, approximately 22 nucleotides in length, which anneal to mRNA target genes, principally through partial complementarity to the 3'-untranslated region of target genes, where they repress protein translation or promote mRNA decay. Aberrant miRNA expression is a hallmark of human tumours and across B-cell lymphomas, deregulated miRNAs have been identified due to their impact on B-cell differentiation, and on established cancer pathways. A number of candidate and genome-wide studies have been performed identifying novel miRNAs and those that are casually implicated in other B-cell lymphomas, such as miR-155, miR-21 and miR-34a[105].
As briefly mentioned previously, several candidate miRNAs have been defined based on their proximity to the minimal deletion region on 7q. Whilst based on limited cohorts and traditional molecular approaches, miRNAs including miR-593, miR-129, miR-182, miR-96, miR-183, miR-335, miR-29a and miR-29b-1 have been shown to be under-expressed in the presence of 7q deletions[106,107]. Under-expression of miR-29a and miR-29b- on 7q, has putative roles in immune regulation, cell proliferation and B-cell differentiation. In particular a key miR-29 target is the TCL1A gene, a gene that is over-expressed in SMZL[92], where miRNA binding deactivates the oncogenic function of TCL1A. Several of these other 7q miRNAs may also contribute to disease biology through the dysregulation of target gene expression and function; miR-129 and miR-335 have been shown to target BCL2, RB1 and BCL-w[108,109]. Beyond miRNAs located on 7q, genome-wide studies have identified a spectrum of miRNAs, some of which target disease-relevant genes and are associated with clinical outcome. Key studies have compared miRNA expression in SMZL vs. other B-cell lymphoma sub-types, or by using normal/reactive spleen samples[110,111]. Bouteloup et al.[111] demonstrated that reduced miR-29a and elevated miR-21 levels correlate with disease aggressiveness. Peveling-Oberhag et al.[112] found miR-26b to be differentially expressed between SMZL arising in Hepatitis-C (HCV)+ vs. HCV- patients, though it remains unknown to what extent miR-26b expression is associated with SMZL pathophysiology or HCV infection. However, there is only limited concordance between these studies, perhaps reflecting biological disease heterogeneity, but also predicated on experimental differences, such as cohort size, experimental design, technology of choice and statistical analysis. Clearly, further investigations are needed to identify and functionally validate causative miRNAs involved in SMZL pathogenesis.
THE CLINICAL UTILITY OF MOLECULAR LESIONS IN SMZL
Previous sections of this review have highlighted in SMZL the biased use of the IGHV1-2*04 allele, a cytogenetic and genomic landscape which overlaps that seen in other marginal zone and splenic lymphomas but is distinguished by a higher incidence of 7q deletion, KLF2 and NOTCH2 mutations and a subgroup characterised by high genome-wide promoter methylation. The latter, together with NOTCH2 mutations, unmutated IGHV genes, TP53 abnormalities and a complex karyotype has been reported in one or more studies to have an adverse prognostic significance[24,31,38]. The European Society for Medical Oncology has recently published guidelines on the diagnosis, treatment and follow up of MZ lymphomas and while acknowledging the potential utility of some of the molecular features listed above as biomarkers, none are currently recommended for establishing the diagnosis, assessing prognosis or determining the timing or choice of therapy.
There are a number of reasons for this apparent paradox; firstly there is a lack of routine access to many of the potential novel biomarkers, secondly the rarity of SMZL and the paucity of clinical trial data dictate that studies have been relatively small and retrospective and thirdly, many cohorts have included a large percentage of non-splenectomised cases in whom establishing a definitive diagnosis may be difficult, especially in cases presenting before the diagnostic features of HCL-v and SDRPL were well recognized. Confining cohorts to cases who have undergone splenectomy overcomes this problem but excludes cases with indolent disease not requiring treatment and those treated with other modalities. Nevertheless, preliminary data from ongoing studies suggest that one or more combinations of IGHV1-2*04 usage, deletion of 7q, NOTCH2 and KLF2 mutations and aberrant methylation will be sufficient to identify a subgroup of SMZL with clearly defined diagnostic and prognostic features. Whilst the role of minimal residual disease monitoring in the management of SMZL patients remains unclear[113], (epi)genomic lesions may have utility to track treatment response, perhaps in conjunction of the use of positron emission tomography scans.
A further benefit of genomic analyses demonstrating the importance of dysregulated NF-κB signalling, partially mediated through increased BCR signalling has been able to provide the rationale for clinical trials of BTK and PI3K inhibitors in all 3 subtypes of MZL. Table 3 summarises response data for patients with SMZL who were refractory to, or relapsed after, one or more prior therapies, usually including an anti-CD20 antibody[114-118]. Overall response rates of 50%-65% are comparable to those seen in other MZL subtypes. It is not yet reported whether primary acquired resistance to these agents is associated with mutations downstream of the PI3K and BCR pathways but in vitro studies using MZL cell lines have demonstrated synergy between copanlisib and the BCL2 inhibitor venetoclax[119], and that acquired copanlisib resistance induced by prolonged exposure, was associated with decreased sensitivity to other PI3K inhibitors and to ibrutinib and with upregulation of multiple signaling pathways[120].
Overall response rates to novel signalling inhibitor in phase 2 studies of relapsed/refractory patients with SMZL. *1 patient was unevaluable
| Phase 2 study | Novel agent | Activity | No. of patients | ORR % |
| PCYC 1121 | Ibrutinib | BTKi | 14 | 62 |
| MAGNOLIA | Zanabrutinib | BTKi | 12 | 50 |
| CHRONOS-1 | Copanlisib | Pan Class 1 PI3Ki | 4 | 75* |
| CITADEL | Parsaclisib | PI3K delta i | 35 | 63 |
| UTX-TGR | Umbralisib | PIK3 delta + CK1 i | 69 MZL (8 SMZL) | 55 (whole cohort) |
FUTURE PERSPECTIVES
Our understanding of the molecular basis of many mature B-cell tumour types has deepened over recent decades, principally through the systematic application of novel technologies with ever increasing resolution and throughput. This information has revolutionized our understanding of the most prevalent mature B-cell lymphomas, yielding novel biological pathways and mechanisms, the ultimate result being improvements in patient stratification, management, and outcomes. In CLL and DLBCL, these studies have included several thousand patients, studies with WGS/WES, complemented with expansive transcriptomic and epigenomic datasets, and distilled with sophisticated computational approaches. Our understanding of SMZL lags far behind and will require significant investment to realise the translational potential of a granular understanding of the SMZL genome and its regulation. The next section will outline key areas for future research focus (summarised in Figure 5), culminating with an overview of two ongoing projects that hope to answer many of these questions.
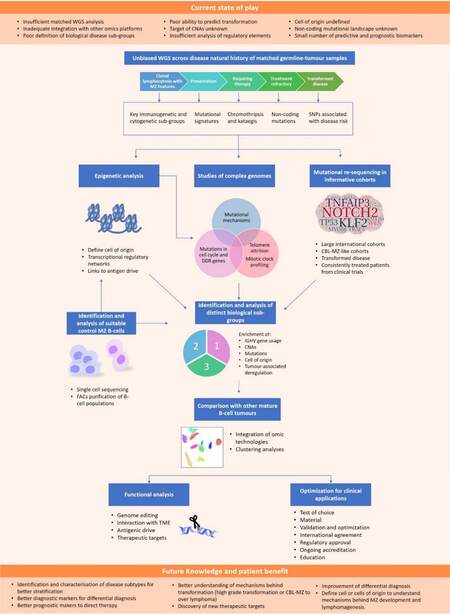
Figure 5. Current state of SMZL knowledge, proposed strategies, and future perspectives. The upper orange box highlights current knowledge in SMZL. The diagram outlines key areas for future research starting with unbiased whole genome sequencing of matched germline-tumour samples across the disease history. Each time point will allow for the identification of key immunogenetic and cytogenetic subgroups, as well as mutational signatures. Whole genome sequencing is essential for the identification of non-coding mutations, and with large collaborative cohorts enabling the identification of SNPs associated with disease risk. This unbiased approach will form the basis for more targeted studies, analysing additional contributory factors such as epigenome of SMZL and telomere attrition mechanisms. The identification and analysis of suitable control MZ B-cells will be important work that will feed into wider studies, particularly epigenetic studies and determining the cell of origin. Synthesis of new information will allow the identification of biological sub-groups and direct downstream functional analyses. The ultimate goal is to translate newly acquired knowledge of the underlying molecular mechanism of SMZL for direct patient benefit: to improve differential diagnosis and aid in the discovery of novel therapeutic targets. SMZL: Splenic marginal zone lymphoma; MZ: marginal zone; CNAs: copy number alterations; WGS: whole genome sequencing; CBL-MZ: clonal B cell lymphocytosis of marginal zone origin; IGHV: immunoglobulin heavy chain variable region; FACS: fluorescence-activated cell sorting; TME: tumour microenvironment.
Key research questions
The research community needs to continue to collaborate, creating bio-banked resources, with high-quality fresh-frozen tumour material and matched normal cells. Currently, the literature does not include a single patient with genome-wide somatic mutational data. Kiel et al.[54] reported WGS without the analysis of matched germ-line material, precluding a meaningful analysis of the presence of non-coding somatic variation and the documentation of underlying molecular mechanisms, such as the presence of key mutational signatures and regions of chromothripsis and kataegis. There is a critical need to analyse germ-line material to identify SNPs associated with disease risk, and the profiling of sequential samples from SMZL patients as their disease evolves, at pre-treatment, and with the development of refractory disease and ultimately transformation. There might also be utility in using circulating free DNA as a non-invasive approach in the management of patients and in the identification of mutations as has been done in other lymphomas[121,122].
Consequently, we should persist in our approach to extend our DNA methylation profiling of high-quality, purified SMZL material from large clinically annotated cohorts reflecting the clinical and biological heterogeneity of the disease, using high-resolution microarrays and reduced representation bisulfite sequencing approaches. Parallel analysis of expansive published data sets and the detailed fluorescence-activated cell sorting purification of additional normal B-cell populations that reflect both the maturation state and the MZ origin of the disease will be critical. This will likely identify further disease subgroups, extending the important study from Arribas et al.[25], which identified the PRC2 epitype using low-resolution array-based promoter DNA methylation and expression profiling. Moreover, a comparison between the methylome of SMZL and other mature B-cell neoplasms, will likely provide valuable information with utility for disease classification, particularly for patients that are currently difficult to precisely diagnose. DNA methylation also has potential in identifying the proliferative history of a tumour cell, as passive accumulation of DNA methylation in repressed regions without detectable function is a feature that can be used as a clock of cellular proliferation[95,123].
Experiments focusing on complex karyotype SMZL cases, particularly given their high frequency, will provide novel biological and clinically relevant information, as complexity points to a key dysregulation of appropriate cell cycle control or DNA damage response pathways in these patients, and has been linked to poor survival in SMZL[31] and in other mature B-cell lymphomas[124-126], including in the context of novel targeted therapies[127,128]. Systematic analysis of the complex landscape of the SMZL genome, with high-density arrays or WGS, would provide a more granular view of the levels and types of complexity that define these patients, and would help clarify disease-specific definition of complexity, which would both aid in appropriate patient prognostication, but also identify key cases for functional analysis; particularly those exhibiting genomic complexity in the absence of established drivers, such as TP53 or ATM disfunction.
Given its intricate association with genomic complexity another attractive approach is the analysis of telomere structure and dynamics. Acute telomere attrition leads to the uncapping of chromatid ends, and in normal tissues results in the activation of senescence checkpoints, with a key pre-malignant tumour-suppressor function. Telomere attrition leads to intra- and inter-chromosomal end fusions, the formation of dicentric chromosomes with consequential breakage during anaphase, and genomic complexity, through the mechanisms of breakage-fusion-bridge formation[129]. The majority of human tumours exhibit eroded telomere length, compared to the corresponding normal tissue[130]. In line with this, several mature B-cell tumour cohorts, particularly CLL, have been shown to contain a proportion of patients with the shortest telomeres, which are more likely to exhibit poor risk-genomic lesions, have unmutated IGHV genes, and harbour a complex genome and significantly poorer clinical outcome[131,132]. However, to date, preliminary telomere length analysis of SMZL has only been published in abstract form showing an enrichment of short telomeres in patients with progressive disease[133]. This approach should be extended to large patient cohorts, and is likely to be highly informative, particularly given the aforenoted enrichment of cases with karyotypic complexity and the significant clinical heterogeneity that exists. There are a number of approaches to quantify telomere length, but one likely approach would be a PCR-based approach, such as the MMQ-PCR assay[134], as this is highly scalable, cost effective and requires a small amount of DNA for analysis. However, given that MMQ-PCR quantifies only a mean telomere length, the single telomere length amplification assay would be the ideal choice, as this allows the quantification of telomere length for a single chromosome in a single cell[135]. There are also approaches being developed such as TeSLA and TCA, both of which employ high-resolution DNA imaging technologies[136,137].
Ongoing studies
Whilst it is clear that SMZL research remains in its infancy, a close and collaborative network of clinicians and scientists are continuing to study the disease. Two ongoing projects are particularly exciting and promise to significantly advance our biological understanding of the disease. Firstly, the IELSG46 (NCT02945319) is an observational study, led by Professor Davide Rossi, which aims to develop and validate an accurate prognostication model using integrated molecular profiling of > 300 treatment naive SMZL patients, where diagnosis is based on spleen histology and > 5 years of follow-up is available on all patients. The initial plans were to perform targeted sequencing to detect somatic gene mutations and permit IGHV analysis and FISH to define 7q deletion status, culminating with complex statistical methods to develop hierarchical models to predict overall survival. However, the project has now developed to include the genome wide analysis of mutations, CNAs and gene expression. This exciting multicentre study is ongoing and has only been presented in abstract form[138,139]. In 2019, the consortium presented their early work. Profiling a cohort of 382 SMZL patients, the authors define four molecular clusters (MC) with distinct outcomes, employing machine learning approaches. MC1 and 2 were driven by KLF2 and NOTCH2 mutations, respectively, and enriched for IGHV1-2*04 and deletion of 7q. MC3 and 4 were defined by KMT2D and TP53/ATM mutations, respectively. Clinical correlations demonstrated that MC1 and MC2 were those that exhibited inferior survival. The authors also reported a more restricted analysis of gene expression data suggesting that patients can be clustered based on transcriptomic signatures. This study, and a host of consequent manuscripts, will be truly important in defining the prognostic landscape of the disease, and will be an important step towards identifying patients for precision medicine approaches.
Our group is also coordinating a large international study, distinct from IELSG46, focusing on detailed DNA methylation profiling of SMZL cases and positioning these cases within a framework including a spectrum of normal B-cell sub-sets and a myriad of other mature B-cell tumours. These data will then be integrated with genome-wide mutational and CNA data, telomere dynamics, and for key biological subgroups, the proliferative history based on methylation arrays, transcriptomic and chromatin accessibility maps. The aims are to: (1) further characterize the epigenetic landscape of SMZL, and define new patient subgroups based on state-of-the-art approaches; (2) define patient subgroups based on their relationship to normal B-cell populations, providing insights into the potential cell-of-origin of key patient subgroups; (3) compare SMZL to the DNA methylation patterns of other mature B-cell tumours, detailing approaches that might aid in differential diagnosis; and (4) compare CBL-MZ to SMZL and aligned conditions, allowing more accurate disease classification and improved prediction of those CBL-MZ cases destined to progress to overt lymphoma. These are two examples of key data sets that will emerge in the literature in the coming months and should advance our understanding of the molecular lesions that define the disease and provide further support of their clinical utility.
CONCLUSIONS
Over recent years, technological advances have provided exponential increases in sensitivity and specificity, thereby transforming our understanding of many of the more prevalent mature B-cell tumours, providing compelling evidence of their clinical utility and facilitating the development of cost effective diagnostic procedures with high accuracy. In SMZL, the picture is less clear. Most cases of SMZL remain indolent with protracted overall survival, but biomarkers are needed to help define the third of patients that exhibit aggressive disease with the possibility of transformation to a more lethal lymphoma. The work outlined herein has improved our understanding of the biological basis of SMZL, with key discoveries being: (1) a highly restricted IGHV gene repertoire, including selective usage of the IGHV1-2*04 allele in 30% of cases; (2) recurrent CNAs (deletion of 7q being the most frequent) and somatic mutations, the latter of which appear to cluster within biologically relevant pathways, such as Notch, NF-κB, BCR and TLR signalling; and (3) a epigenetically-defined disease sub-group with PRC2 activation.
These observations are translationally important with clear clinical utility and will help with the diagnosis and risk-adapted patient stratification in the coming years if the appropriate testing procedures can be agreed upon. This is a challenging aim, needing the most appropriate assays that identify the most clinically informative immunogenetic and (epi)genomic information. In our lab, we employ two resequencing panels, one for IGHV analysis and another to identify gene mutations associated with survival (KLF2, NOTCH2, TP53, TNFAIP3) and those that might provide information on differential diagnosis [WM (MYD88), HCL (BRAF), CLL (NOTCH1, SF3B1), NMZL (PTPRD), HCLv (MAP2K1)]. We spike our panel design with a backbone of genome-wide probes (200 kb resolution) for CNA identification. Our view is that this approach is a suitable balance between genome coverage and the sequencing depth to identify sub-clonal mutations, with appropriate reagent and sequencing costs, and the time and computational resources required for analysis. As our understanding of the genome increases, these gene panels could be expanded with further prognostically relevant lesions, those that might be therapeutic targets, and those that might help monitor response. Ultimately, it might be that comprehensive multi-omics analysis (including epigenetic profiling) will most accurately identify patients for precision medicine, but it will need to be offset by financial savings through selection of the most appropriate treatment choice. Independent of the technical approach, each molecular marker would need to be validated across multiple discovery and validation cohorts, in retrospective historical studies and in prospective clinical trials, resulting in a robust and reproducible association with a disease outcome. Assays would then need to be analytically and clinically validated, before international harmonization, regulatory approval, and ongoing assessment through accreditation. Guidelines for interpretation and reporting would be required, and training would be needed to facilitate appropriate clinical referral and accurate communication with patients.
DECLARATIONS
Authors’ contributionsPrepared the first draft of the manuscript: Jaramillo Oquendo C, Strefford JC
The assistance: Oscier D
Appraised the manuscript and contributed to the final draft: Jaramillo Oquendo C, Parker H, Oscier D, Ennis S, Gibson J, Strefford JC
Availability of data and materialsNot applicable.
Financial support and sponsorshipJaramillo Oquendo C funded by COLCIENCAS. Parker H is funded by the Kay Kendall Leukaemia Fund (873). Strefford JC received funding from Cancer Research UK.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021
REFERENCES
1. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375-90.
2. Kanellis G, Mollejo M, Montes-Moreno S, et al. Splenic diffuse red pulp small B-cell lymphoma: revision of a series of cases reveals characteristic clinico-pathological features. Haematologica 2010;95:1122-9.
3. Traverse-Glehen A, Baseggio L, Salles G, Coiffier B, Felman P, Berger F. Splenic diffuse red pulp small-B cell lymphoma: toward the emergence of a new lymphoma entity. Discov Med 2012;13:253-65.
4. Xochelli A, Kalpadakis C, Gardiner A, et al. Clonal B-cell lymphocytosis exhibiting immunophenotypic features consistent with a marginal-zone origin: is this a distinct entity? Blood 2014;123:1199-206.
5. Merli M, Bianchi B, Bertù L, et al. Clonal B-cell lymphocytosis with mariginal-zone features: comparison with overt splenic marginal-zone lymphomas in 77 patients from a monocentric series. Blood 2019;134:4017.
6. Chacón JI, Mollejo M, Muñoz E, Algara P, Mateo M, Lopez L, et al. Splenic marginal zone lymphoma: clinical characteristics and prognostic factors in a series of 60 patients. Blood 2002;100:1648-54.
7. Matutes E, Oscier D, Montalban C, et al. Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia 2008;22:487-95.
8. Zucca E, Arcaini L, Buske C, et al. ESMO Guidelines Committee. Electronic address: clinicalguidelines@esmo.org. Marginal zone lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2020;31:17-29.
9. Lumish M, Falchi L, Imber BS, Scordo M, von Keudell G, Joffe E. How we treat mature B-cell neoplasms (indolent B-cell lymphomas). J Hematol Oncol 2021;14:5.
10. Arcaini L, Rossi D, Paulli M. Splenic marginal zone lymphoma: from genetics to management. Blood 2016;127:2072-81.
11. Melo JV, Hegde U, Parreira A, Thompson I, Lampert IA, Catovsky D. Splenic B cell lymphoma with circulating villous lymphocytes: differential diagnosis of B cell leukaemias with large spleens. J Clin Pathol 1987;40:642-51.
12. Mulligan SP, Matutes E, Dearden C, Catovsky D. Splenic lymphoma with villous lymphocytes: natural history and response to therapy in 50 cases. Br J Haematol 1991;78:206-9.
13. Schmid C, Kirkham N, Diss T, Isaacson PG. Splenic marginal zone cell lymphoma. Am J Surg Pathol 1992;16:455-66.
14. Baldini L, Fracchiolla N, Cro L, et al. Frequent p53 gene involvement in splenic B-cell leukemia/lymphomas of possible marginal zone origin. Blood 1994;84:270-8.
15. Zhu D, Oscier DG, Stevenson FK. Splenic lymphoma with villous lymphocytes involves B cells with extensively mutated Ig heavy chain variable region genes. Blood 1995;85:1603-7.
16. Mollejo M, Menárguez J, Lloret E, et al. Splenic marginal zone lymphoma: a distinctive type of low-grade B-cell lymphoma. A clinicopathological study of 13 cases. Am J Surg Pathol 1995;19:1146-57.
17. Hermine O, Lefrère F, Bronowicki JP, et al. Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection. N Engl J Med 2002;347:89-94.
18. Novak U, Rinaldi A, Kwee I, et al. The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 2009;113:4918-21.
19. Oscier DG, Matutes E, Gardiner A, et al. Cytogenetic studies in splenic lymphoma with villous lymphocytes. Br J Haematol 1993;85:487-91.
20. Bikos V, Darzentas N, Hadzidimitriou A, et al. Over 30% of patients with splenic marginal zone lymphoma express the same immunoglobulin heavy variable gene: ontogenetic implications. Leukemia 2012;26:1638-46.
21. Xochelli A, Bikos V, Polychronidou E, et al. Disease-biased and shared characteristics of the immunoglobulin gene repertoires in marginal zone B cell lymphoproliferations. J Pathol 2019;247:416-21.
22. Bikos V, Karypidou M, Stalika E, et al. An Immunogenetic Signature of Ongoing Antigen Interactions in Splenic Marginal Zone Lymphoma Expressing IGHV1-2*04 Receptors. Clin Cancer Res 2016;22:2032-40.
23. Clipson A, Wang M, de Leval L, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia 2015;29:1177-85.
24. Parry M, Rose-Zerilli MJ, Ljungström V, et al. Genetics and Prognostication in Splenic Marginal Zone Lymphoma: Revelations from Deep Sequencing. Clin Cancer Res 2015;21:4174-83.
25. Arribas AJ, Rinaldi A, Mensah AA, et al. DNA methylation profiling identifies two splenic marginal zone lymphoma subgroups with different clinical and genetic features. Blood 2015;125:1922-31.
26. Arribas AJ, Bertoni F. Methylation patterns in marginal zone lymphoma. Best Pract Res Clin Haematol 2017;30:24-31.
27. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig VH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848-54.
28. Damle RN, Wasil T, Fais F, et al. Ig V Gene Mutation Status and CD38 Expression As Novel Prognostic Indicators in Chronic Lymphocytic Leukemia. Blood 1999;94:1840-7.
29. Rinaldi A, Mian M, Chigrinova E, et al. Genome-wide DNA profiling of marginal zone lymphomas identifies subtype-specific lesions with an impact on the clinical outcome. Blood 2011;117:1595-604.
30. Arcaini L, Zibellini S, Passamonti F, et al. Splenic marginal zone lymphoma: Clinical clustering of immunoglobulin heavy chain repertoires. Blood Cells Mol Dis 2009;42:286-91.
31. Salido M, Baró C, Oscier D, et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: a multicenter study of the Splenic B-Cell Lymphoma Group. Blood 2010;116:1479-88.
32. Rossi D, Deaglio S, Dominguez-Sola D, et al. Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood 2011;118:4930-4.
33. Piva R, Deaglio S, Famà R, et al. The Krüppel-like factor 2 transcription factor gene is recurrently mutated in splenic marginal zone lymphoma. Leukemia 2015;29:503-7.
34. Davoli T, Xu AW, Mengwasser KE, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013;155:948-62.
35. Sack LM, Davoli T, Li MZ, et al. Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell 2018;173:499-514.e23.
36. Watkins AJ, Huang Y, Ye H, et al. Splenic marginal zone lymphoma: characterization of 7q deletion and its value in diagnosis. J Pathol 2010;220:461-74.
37. Algara P, Mateo MS, Sanchez-Beato M, et al. Analysis of the IgV(H) somatic mutations in splenic marginal zone lymphoma defines a group of unmutated cases with frequent 7q deletion and adverse clinical course. Blood 2002;99:1299-304.
38. Fresquet V, Robles EF, Parker A, et al. High-throughput sequencing analysis of the chromosome 7q32 deletion reveals IRF5 as a potential tumour suppressor in splenic marginal-zone lymphoma. Br J Haematol 2012;158:712-26.
39. Hosono N, Makishima H, Mahfouz R, et al. Recurrent genetic defects on chromosome 5q in myeloid neoplasms. Oncotarget 2017;8:6483-95.
40. Venugopal S, Mascarenhas J, Steensma DP. Loss of 5q in myeloid malignancies - A gain in understanding of biological and clinical consequences. Blood Rev 2021;46:100735.
41. Dierlamm J, Rosenberg C, Stul M, et al. Characteristic pattern of chromosomal gains and losses in marginal zone B cell lymphoma detected by comparative genomic hybridization. Leukemia 1997;11:747-58.
42. Dierlamm J, Michaux L, Wlodarska I, et al. Trisomy 3 in marginal zone B-cell lymphoma: a study based on cytogenetic analysis and fluorescence in situ hybridization. Br J Haematol 1996;93:242-9.
43. Hernández JM, García JL, Gutiérrez NC, et al. Novel genomic imbalances in B-Cell splenic marginal zone lymphomas revealed by comparative genomic hybridization and cytogenetics. Am J Pathol 2001;158:1843-50.
44. Solé F, Woessner S, Florensa L, et al. Frequent involvement of chromosomes 1, 3, 7 and 8 in splenic marginal zone B-cell lymphoma. Br J Haematol 1997;98:446-9.
45. Troussard X, Mauvieux L, Radford-Weiss I, et al. Genetic analysis of splenic lymphoma with villous lymphocytes: a Groupe Français d'Hématologie Cellulaire (GFHC) study. Br J Haematol 1998;101:712-21.
46. Robledo C, García JL, Benito R, et al. Molecular characterization of the region 7q22.1 in splenic marginal zone lymphomas. PLoS One 2011;6:e24939.
47. Gailllard B, Cornillet-Lefebvre P, Le QH, et al. Groupe Francophone de Cytogénétique Hématologique. Clinical and biological features of B-cell neoplasms with CDK6 translocations: an association with a subgroup of splenic marginal zone lymphomas displaying frequent CD5 expression, prolymphocytic cells, and TP53 abnormalities. Br J Haematol 2021;193:72-82.
48. Hoang PH, Dobbins SE, Cornish AJ, et al. Whole-genome sequencing of multiple myeloma reveals oncogenic pathways are targeted somatically through multiple mechanisms. Leukemia 2018;32:2459-70.
49. Burns A, Alsolami R, Becq J, et al. Whole-genome sequencing of chronic lymphocytic leukaemia reveals distinct differences in the mutational landscape between IgHVmut and IgHVunmut subgroups. Leukemia 2018;32:332-42.
50. Arthur SE, Jiang A, Grande BM, et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat Commun 2018;9:4001.
51. Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 2011;44:47-52.
52. Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011;475:101-5.
53. Oquendo C, Parker H, Oscier D, Ennis S, Gibson J, Strefford JC. Systematic Review of Somatic Mutations in Splenic Marginal Zone Lymphoma. Sci Rep 2019;9:10444.
54. Kiel MJ, Velusamy T, Betz BL, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med 2012;209:1553-65.
55. Luijten MNH, Lee JXT, Crasta KC. Mutational game changer: Chromothripsis and its emerging relevance to cancer. Mutat Res 2018;777:29-51.
56. Parry M, Rose-Zerilli MJ, Gibson J, et al. Whole exome sequencing identifies novel recurrently mutated genes in patients with splenic marginal zone lymphoma. PLoS One 2013;8:e83244.
57. Rossi D, Trifonov V, Fangazio M, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med 2012;209:1537-51.
58. Campos-Martín Y, Martínez N, Martínez-López A, et al. Clinical and diagnostic relevance of NOTCH2-and KLF2-mutations in splenic marginal zone lymphoma. Haematologica 2017;102:e310-2.
59. Jallades L, Baseggio L, Sujobert P, et al. Exome sequencing identifies recurrent BCOR alterations and the absence of KLF2, TNFAIP3 and MYD88 mutations in splenic diffuse red pulp small B-cell lymphoma. Haematologica 2017;102:1758-66.
60. Feinberg MW, Lin Z, Fisch S, Jain MK. An emerging role for Krüppel-like factors in vascular biology. Trends Cardiovasc Med 2004;14:241-6.
61. Dang DT, Pevsner J, Yang VW. The biology of the mammalian Krüppel-like family of transcription factors. Int J Biochem Cell Biol 2000;32:1103-21.
62. Hart GT, Wang X, Hogquist KA, Jameson SC. Krüppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc Natl Acad Sci U S A 2011;108:716-21.
63. Winkelmann R, Sandrock L, Porstner M, et al. B cell homeostasis and plasma cell homing controlled by Krüppel-like factor 2. Proc Natl Acad Sci U S A 2011;108:710-5.
64. Hoek KL, Gordy LE, Collins PL, et al. Follicular B cell trafficking within the spleen actively restricts humoral immune responses. Immunity 2010;33:254-65.
65. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47:D886-94.
66. Tanigaki K, Han H, Yamamoto N, et al. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat Immunol 2002;3:443-50.
67. Saito T, Chiba S, Ichikawa M, et al. Notch2 Is Preferentially Expressed in Mature B Cells and Indispensable for Marginal Zone B Lineage Development. Immunity 2003;18:675-85.
68. Arruga F, Vaisitti T, Deaglio S. The NOTCH Pathway and Its Mutations in Mature B Cell Malignancies. Front Oncol 2018;8:550.
69. Moran ST, Cariappa A, Liu H, et al. Synergism between NF-kappa B1/p50 and Notch2 during the development of marginal zone B lymphocytes. J Immunol 2007;179:195-200.
71. Radtke F, Wilson A, MacDonald HR. Notch signaling in T- and B-cell development. Curr Opin Immunol 2004;16:174-9.
72. Martínez N, Almaraz C, Vaqué JP, et al. Whole-exome sequencing in splenic marginal zone lymphoma reveals mutations in genes involved in marginal zone differentiation. Leukemia 2014;28:1334-40.
73. Shanmugam V, Craig JW, Hilton LK, et al. Notch activation is pervasive in SMZL and uncommon in DLBCL: implications for Notch signaling in B-cell tumors. Blood Adv 2021;5:71-83.
74. Hampel F, Ehrenberg S, Hojer C, et al. CD19-independent instruction of murine marginal zone B-cell development by constitutive Notch2 signaling. Blood 2011;118:6321-31.
75. Fabbri G, Holmes AB, Viganotti M, et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2017;114:E2911-9.
77. Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol 2009;9:767-77.
78. Spina V, Rossi D. NF-κB deregulation in splenic marginal zone lymphoma. Semin Cancer Biol 2016;39:61-7.
79. Lim KH, Yang Y, Staudt LM. Pathogenetic importance and therapeutic implications of NF-κB in lymphoid malignancies. Immunol Rev 2012;246:359-78.
80. Yan Q, Huang Y, Watkins AJ, et al. BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica 2012;97:595-8.
81. Pillonel V, Juskevicius D, Ng CKY, et al. High-throughput sequencing of nodal marginal zone lymphomas identifies recurrent BRAF mutations. Leukemia 2018;32:2412-26.
82. Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011;470:115-9.
83. Lunning MA, Green MR. Mutation of chromatin modifiers; an emerging hallmark of germinal center B-cell lymphomas. Blood Cancer J 2015;5:e361.
84. Spina V, Khiabanian H, Messina M, et al. The genetics of nodal marginal zone lymphoma. Blood 2016;128:1362-73.
86. Zhang J, Dominguez-Sola D, Hussein S, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 2015;21:1190-8.
87. Green MR, Kihira S, Liu CL, et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A 2015;112:E1116-25.
88. Okosun J, Bödör C, Wang J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 2014;46:176-81.
89. Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 2004;432:635-9.
90. Han L, Madan V, Mayakonda A, et al. Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia 2019;33:2291-305.
91. Trøen G, Nygaard V, Jenssen T, et al. Constitutive expression of the AP-1 transcription factors c-jun, junD, junB, and c-fos and the marginal zone B-Cell transcription factor notch2 in splenic marginal zone lymphoma. J Mol Diagn 2004;6:297-307.
92. Ruiz-Ballesteros E, Mollejo M, Rodriguez A, et al. Splenic marginal zone lymphoma: proposal of new diagnostic and prognostic markers identified after tissue and cDNA microarray analysis. Blood 2005;106:1831-8.
93. Navarro A, Clot G, Martínez-Trillos A, et al. Improved classification of leukemic B-cell lymphoproliferative disorders using a transcriptional and genetic classifier. Haematologica 2017;102:e360-3.
94. Robinson JE, Greiner TC, Bouska AC, Iqbal J, Cutucache CE. Identification of a Splenic Marginal Zone Lymphoma Signature: Preliminary Findings With Diagnostic Potential. Front Oncol 2020;10:640.
95. Duran-ferrer M, Clot G, Nadeu F, et al. The proliferative history shapes the DNA methylome of B-cell tumors and predicts clinical outcome. Nat Cancer 2020;1:1066-81.
96. Queirós AC, Beekman R, Vilarrasa-Blasi R, et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell 2016;30:806-21.
97. Kulis M, Merkel A, Heath S, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet 2015;47:746-56.
98. Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet 2012;44:1236-42.
99. Lee ST, Muench MO, Fomin ME, et al. Epigenetic remodeling in B-cell acute lymphoblastic leukemia occurs in two tracks and employs embryonic stem cell-like signatures. Nucleic Acids Res 2015;43:2590-602.
100. Su IH, Basavaraj A, Krutchinsky AN, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 2003;4:124-31.
101. Akasaka T, Tsuji K, Kawahira H, et al. The Role of mel-18, a Mammalian Polycomb Group Gene, during IL-7-Dependent Proliferation of Lymphocyte Precursors. Immunity 1997;7:135-46.
102. Oakes CC, Seifert M, Assenov Y, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet 2016;48:253-64.
103. Ziller MJ, Gu H, Müller F, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013;500:477-81.
104. Jiang Y, Hatzi K, Shaknovich R. Mechanisms of epigenetic deregulation in lymphoid neoplasms. Blood 2013;121:4271-9.
105. Solé C, Arnaiz E, Lawrie CH. MicroRNAs as Biomarkers of B-cell Lymphoma. Biomark Insights 2018;13:1177271918806840.
106. Watkins AJ, Hamoudi RA, Zeng N, et al. An integrated genomic and expression analysis of 7q deletion in splenic marginal zone lymphoma. PLoS One 2012;7:e44997.
107. Ruiz-Ballesteros E, Mollejo M, Mateo M, Algara P, Martínez P, Piris MA. MicroRNA losses in the frequently deleted region of 7q in SMZL. Leukemia 2007;21:2547-9.
108. Karaayvaz M, Zhai H, Ju J. miR-129 promotes apoptosis and enhances chemosensitivity to 5-fluorouracil in colorectal cancer. Cell Death Dis 2013;4:e659.
109. Scarola M, Schoeftner S, Schneider C, Benetti R. miR-335 directly targets Rb1 (pRb/p105) in a proximal connection to p53-dependent stress response. Cancer Res 2010;70:6925-33.
110. Di Lisio L, Sánchez-Beato M, Gómez-López G, et al. MicroRNA signatures in B-cell lymphomas. Blood Cancer J 2012;2:e57.
111. Bouteloup M, Verney A, Rachinel N, et al. MicroRNA expression profile in splenic marginal zone lymphoma. Br J Haematol 2012;156:279-81.
112. Peveling-Oberhag J, Crisman G, Schmidt A, et al. Dysregulation of global microRNA expression in splenic marginal zone lymphoma and influence of chronic hepatitis C virus infection. Leukemia 2012;26:1654-62.
113. Cervetti G, Galimberti S, Sordi E, et al. Significant efficacy of 2-CdA with or without rituximab in the treatment of splenic marginal zone lymphoma (SMZL). Ann Oncol 2010;21:851-4.
114. Phillips TJ, Corradini P, Gurion R, et al. Phase 2 study evaluating the efficacy and safety of parsaclisib in patients with relapsed or refractory marginal zone lymphoma (CITADEL-204). Blood 2020;136:27-8.
115. Opat S, Tedeschi A, Linton K, et al. Efficacy and safety of zanubrutinib in patients with relapsed/refractory marginal zone lymphoma: initial results of the MAGNOLIA (BGB-3111-214) trial. Blood 2020;136:28-30.
116. Zinzani P, Samaniego F, Jurczak W, et al. Umbralisib monotherapy demonstrates efficacy and safety in patients with relapsed/refractory marginal zone lymphoma: a multicenter, open-label, registration directed phase 2 study. Hematol Oncol 2019;37:182-3.
117. Noy A, de Vos S, Coleman M, et al. Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: long-term follow-up and biomarker analysis. Blood Adv 2020;4:5773-84.
118. Panayiotidis P, Follows GA, Mollica L, et al. Efficacy and safety of copanlisib in patients with relapsed or refractory marginal zone lymphoma. Blood Adv 2021;5:823-8.
119. Tarantelli C, Lange M, Gaudio E, et al. Copanlisib synergizes with conventional and targeted agents including venetoclax in B- and T-cell lymphoma models. Blood Adv 2020;4:819-29.
120. Arribas AJ, Napoli S, Cascione L, et al. Abstract PO-46: Mechanisms of resistance to the PI3K inhibitor copanlisib in marginal zone lymphoma. Blood Cancer Discovery 2020; doi: 10.1158/2643-3249.
121. Andrade-campos MM, Salar A, Sanchez-gonzalez B, et al. Assessment of cell-free DNA (cfDNA) in 221 patients with lymphoproliferative malignancies at diagnosis and during follow-up. Blood 2019;134:492.
122. Rossi D, Diop F, Spaccarotella E, et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017;129:1947-57.
123. Zhou W, Dinh HQ, Ramjan Z, et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat Genet 2018;50:591-602.
124. Le Bris Y, Struski S, Guièze R, et al. Major prognostic value of complex karyotype in addition to TP53 and IGHV mutational status in first-line chronic lymphocytic leukemia. Hematol Oncol 2017;35:664-70.
125. Thompson PA, O'Brien SM, Wierda WG, et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer 2015;121:3612-21.
126. Greenwell IB, Staton AD, Lee MJ, et al. Complex karyotype in patients with mantle cell lymphoma predicts inferior survival and poor response to intensive induction therapy. Cancer 2018;124:2306-15.
127. Anderson MA, Tam C, Lew TE, et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood 2017;129:3362-70.
128. Brown JR, Hillmen P, O'Brien S, et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia 2018;32:83-91.
129. Gisselsson D, Pettersson L, Höglund M, et al. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci U S A 2000;97:5357-62.
130. Barthel FP, Wei W, Tang M, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet 2017;49:349-57.
131. Rampazzo E, Bonaldi L, Trentin L, et al. Telomere length and telomerase levels delineate subgroups of B-cell chronic lymphocytic leukemia with different biological characteristics and clinical outcomes. Haematologica 2012;97:56-63.
132. Strefford JC, Kadalayil L, Forster J, et al. Telomere length predicts progression and overall survival in chronic lymphocytic leukemia: data from the UK LRF CLL4 trial. Leukemia 2015;29:2411-4.
133. Parker AE, Snell K, Davis Z, Oscier DG. Telomere length by Tel-PCR in B cell malignancies. Blood 2005;106:2955.
135. Baird DM, Rowson J, Wynford-Thomas D, Kipling D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat Genet 2003;33:203-7.
136. Lai TP, Zhang N, Noh J, et al. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat Commun 2017;8:1356.
137. Kahl VFS, Allen JAM, Nelson CB, et al. Telomere Length Measurement by Molecular Combing. Front Cell Dev Biol 2020;8:493.
138. Bruscaggin A, Mollejo M, Tapia G, et al. Multi-omics landscape of splenic marginal zone lymphoma (SMZL) - interim analysis of IELSG46 study. Hematol Oncol 2019;37:181-2.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Oquendo CJ, Parker H, Oscier D, Ennis S, Gibson J, Strefford JC. The (epi)genomic landscape of splenic marginal zone lymphoma, biological implications, clinical utility, and future questions. J Transl Genet Genom 2021;5:89-111. http://dx.doi.org/10.20517/jtgg.2021.04
AMA Style
Oquendo CJ, Parker H, Oscier D, Ennis S, Gibson J, Strefford JC. The (epi)genomic landscape of splenic marginal zone lymphoma, biological implications, clinical utility, and future questions. Journal of Translational Genetics and Genomics. 2021; 5(2): 89-111. http://dx.doi.org/10.20517/jtgg.2021.04
Chicago/Turabian Style
Oquendo, Carolina Jaramillo, Helen Parker, David Oscier, Sarah Ennis, Jane Gibson, Jonathan C. Strefford. 2021. "The (epi)genomic landscape of splenic marginal zone lymphoma, biological implications, clinical utility, and future questions" Journal of Translational Genetics and Genomics. 5, no.2: 89-111. http://dx.doi.org/10.20517/jtgg.2021.04
ACS Style
Oquendo, CJ.; Parker H.; Oscier D.; Ennis S.; Gibson J.; Strefford JC. The (epi)genomic landscape of splenic marginal zone lymphoma, biological implications, clinical utility, and future questions. J. Transl. Genet. Genom. 2021, 5, 89-111. http://dx.doi.org/10.20517/jtgg.2021.04
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 26 clicks
Cite This Article 26 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.