
Genomics and precision medicine in pediatric acute lymphoblastic leukemia
 0
0Abstract
Acute lymphoblastic leukemia (ALL) is the most frequent malignant disease in the pediatric population, accounting for about 25% of childhood cancers. Drastic therapeutic improvements have been made for pediatric ALL since the early 1960s, marking the most successful treatment paradigm in pediatric oncology. The clinical success derived from refined risk-adapted therapy based on presenting features, cytogenetics and minimal residual disease, prevention of central nervous system relapse, and improvement of supportive care measures. With contemporary therapies, survival of children with ALL now exceeds 90%. However, ALL represents one of leading causes of cancer-related death, as 15%-20% of patients continue to relapse and outcomes post-relapse remain poor. Since the early 2000s, large-scale genomic studies of ALL, greatly facilitated by the advent of next generation sequencing (NGS), have enabled the development of a novel taxonomy for ALL in the molecular era. The access to NGS technologies identifies novel ALL subsets characterized by “driver” oncogenic alterations, previously cryptic on conventional karyotyping methods. With genomic characterization, the group of formerly unclassified B-lineage ALL reduces from 25% to a marginal 5% of ALL. The revised molecular classification of ALL confers prognostic significance and describes the predilection of unfavorable ALL subtypes with increasing age, partially elucidating the worst outcome of adolescents and young adults with ALL. Large-scale genomic analysis also reveals inherited alterations predisposing to ALL occurrence or to different drugs’ sensitivities. Most importantly, the genomic portrait of ALL uncovers novel therapeutic vulnerabilities, paving the way towards precision medicine opportunities in ALL.
Keywords
INTRODUCTION
In the last 60 years, substantial progress has been made in the management of pediatric acute lymphoblastic leukemia (ALL) that translated into meaningful survival improvement. With modern therapies, more than 90% of children with ALL now become long-term survivors[1]. The key factor for this success was the refinement of multi-agent chemotherapy regimens’ delivery via enrollment of thousands of children with ALL onto prospective randomized clinical trials. Results of these clinical trials define the current standard of care and highlight the importance of central nervous system-directed therapy and risk-adapted therapy based on patient’s presenting characteristics, leukemia biology and early response as measured by minimal residual disease (MRD)[2,3]. Clinical characteristics encompassing from age and leukocyte count at diagnosis, leukemia immunophenotype to extramedullary disease involvement, have been first pinpointed as prognostic factors and universally integrated for risk stratification and treatment assignment for childhood ALL. For instance, age ≥ 10 years at diagnosis and presenting leukocyte count ≥ 50 × 109/L [the National Cancer Institute (NCI)-Rome criteria], as well as extramedullary disease and T-cell phenotype classify the patients in the high-risk (HR) group due to lower survival[4]. Sentinel genetic alterations, characterized by chromosomal aneuploidies or rearrangements via conventional cytogenetics, represent an essential component of risk stratification. Some alterations are associated with favorable prognosis, such as high hyperdiploidy (51-65 chromosomes) and ETV6-RUNX1; while other cytogenetic abnormalities including low hypodiploidy (< 40 chromosomes) or near-haploidy, intrachromosomal amplification of chromosome 21 (iAMP21), BCR-ABL1 or Philadelphia chromosome (Ph+), KMT2A (formerly known as MLL) rearrangement (KMT2Ar) and TCF3-HLF confer a worse prognosis[5-7]. Risk-group stratification based on the patient’s clinical and molecular characteristics for tailored therapy intensification has contributed to the dramatic improvement of pediatric ALL’s prognosis over the last 50 years[8]. However, up to 25% of B-lineage ALL (B-ALL) remain unclassified by conventional cytogenetics and is referred to as B-Others[9]. The advent of high-throughput genomic approaches has marked a new paradigm in ALL characterization, revealing a diverse spectrum of subtype-defining alterations that were missed due to their cryptic nature or undetectable by orthogonal methods. These novel alterations can be divided into three different categories: (1) sequence mutations affecting transcription factors (e.g., PAX5 P80R, IKZF1 N159Y); (2) recurrent rearrangement of a single gene with multiple partners (e.g., ZNF384, MEF2D and NUTM1-rearranged ALL); (3) a range of different alterations involving multiples genes within the same molecular group [e.g., PAX5 alterations, DUX4/ERG subtype, ETV6-RUNX1-like ALL, or Philadelphia chromosome-like (Ph-like) ALL][8-11]. Next-generation sequencing (NGS) platforms and large-scale genome-wide studies, especially microarray for copy number alterations (CNAs) and whole-transcriptome analysis, display a high ability to classify new molecular subgroups based on their gene expression profiles (GEP). This approach has unraveled new oncogenic drivers of leukemogenesis. Many of them have been shown to have prognostic and/or therapeutic implications[2,9]. The frequency of each molecular subgroup varies with age; thus partially elucidates the age-related differential outcome in ALL[10]. In this review, we aim to provide an overview of the most recent advances in ALL genomics, and to highlight the prognostic impact and therapeutic opportunities derived from this modern classification. It must be specified that some of these novel entities discussed herein should be considered as provisional. Their prognostic and therapeutic significance will require further validation in large, prospective and uniformly-treated patient cohorts.
MOLECULAR CLASSIFICATION OF ALL
B-ALL
In the early 2000s, Yeoh et al.[12] confirmed that GEP is able to accurately classify known cytogenetic subgroups and sometimes, rectify karyotyping misclassification for the ETV6-RUNX1 subgroup. They also highlighted unique gene expression phenotypes identifying novel subgroups for previously unclassified B-ALL[12]. Furthermore, gene expression clustering correlates with outcome and can be used for prognostic and risk-group classification[13,14]. Herein, we will only emphasize on the recently discovered molecular subgroups, their potential prognostic association and their predilection according to age group. Figure 1 summarizes the B-ALL subgroups with their distribution by age group and a preliminary proposition of risk-group classification.
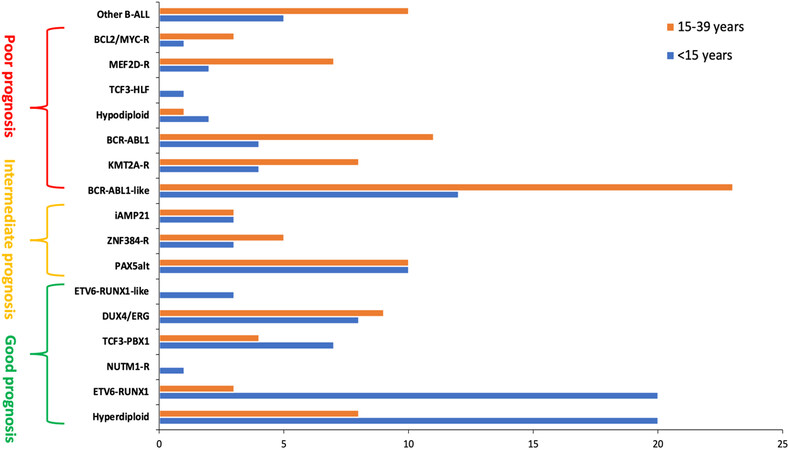
Figure 1. Frequencies of B-lineage acute lymphoblastic leukemia subtypes according to age group. The subtypes are organized by risk-group categories as proposed by the authors based on the available literature. This classification remains provisional as the prognostic significance of rare and novel ALL subtypes can be influenced by treatment strategies and will require further confirmation in larger studies. ALL: Acute lymphoblastic leukemia.
Ph-like ALL
The discovery of Ph-like or BCR-ABL1-like ALL hails from genomic exploration of HR B-ALL[13-17]. This subgroup is defined by an activated kinase gene expression profile, similar to that of Ph+ ALL but missing the canonical BCR-ABL1 fusion. This subset was identified in 2009 by two independent ALL cohorts defined by two distinct gene classifiers that shared only 7 genes in common[15,16]. Interestingly, Ph-like ALL is associated with a worse prognosis than other HR B-ALL and comparable to that of Ph+ ALL[15]. In multivariable analysis, the prognostic significance of Ph-like ALL was retained as an independent adverse outcome biomarker for relapse[18]. This subgroup accounts for ~15% of pediatric B-ALL and increases with age and risk group. In younger children, it represents 10% of standard-risk (SR) ALL and 13% among HR-ALL. The prevalence increases to 21% among adolescents and 27% in young adults and then stabilizes around 20% in older adults after the age of 40[19,20]. Alterations (deletions and inactivating mutations) of IKZF1 or other lymphoid transcription factors (CDKN2A/B, PAX5, ERG, ETV6) and kinase-activating alterations constitute the molecular hallmark of Ph-like ALL[13,14,16,19]. CRLF2 rearrangements (e.g., IGH-CRLF2 and P2RY8-CRLF2), conferring CRLF2 overexpression, comprise half of Ph-like ALL, often harbor concomitant JAK mutations or other JAK-STAT pathway alterations (SH2B3, IL7R) in about 50% of the CRLF2-rearranged cases[19]. CRLF2 rearrangements are associated with a worse prognosis and are more frequent in older children and in people with Hispanic or Native American origin[17]. Overall, more than 90% of Ph-like ALL harbor a myriad of kinase-activating alterations that can be further divided in 2 major categories: (1) alterations activating JAK-STAT signaling pathways, predominantly rearrangements of CRLF2, JAK2 and EPOR; and (2) translocations involving ABL-class genes (ABL1, ABL2, CSF1R, PDGFRA, PDGFRB, LYN). A small number of Ras pathway (KRAS, NRAS, PTPN11, NF1) mutations have been identified; nevertheless, it remains unclear whether these mutations represent pathogenic drivers in Ph-like ALL or only contribute to a similar kinase-activated signature. In addition, some rare fusions involving other tyrosine kinases such as NTRK3, DKGH or FLT3 have been reported in Ph-like ALL[10,19,20]. IKZF1 (gene encoding for the transcription factor IKAROS) intragenic deletions and inactivating mutations are preponderant in kinase-activating leukemia such as Ph+ or Ph-like ALL[14,16,21,22]. IKZF1 alterations are present in up to 70% of Ph+ ALL and Ph-like ALL and confer a worse prognosis compared to their respective counterparts with wild-type IKZF1[19,23]. Nevertheless, the independent negative prognostic impact of IKZF1 deletion in B-ALL is still debatable. IKZF1 was first described as a pejorative marker for the occurrence of relapse[16], but multivariable analyses across different consortia had come to conflicting conclusions. In the Dana Farber Cancer Institute (DFCI) ALL 05-001 study, IKZF1 deletion was associated with poor survival irrespective of the presence of kinase fusion and MRD among 105 NCI HR B-ALL cases[7,23]. However, the results from Children’s Oncology Group (COG) P9905/P9906 trials failed to confirm the prognostic impact of IKZF1 alterations in multivariable analysis when analyzed by risk group[22,24]. The enrichment of IKZF1 alterations in the already known unfavorable Ph-like ALL represent major confounders and contribute to the uncertain prognostic impact of IKZF1. A new category, called IKZF1plus, regrouping IKZF1 deletions that co-occur with CDKN2A, CDKN2B, PAX5 or PAR1 deletion in the absence of ERG deletion could represent a more homogeneous and discriminative subset with high risk of relapse[25].
DUX4-rearranged and ERG-deregulated ALL
Gene expression profiling of HR B-ALL from the COG P9906 cohort discovered a new cluster with an excellent prognosis, contrasting with the overall outcome of HR ALL. This subgroup presents with recurrent intragenic ERG deletion[14], and has been later identified as DUX4-rearranged and ERG-dysregulated (DUX4/ERG) subtype, accounting for 4% to 7% of B-ALL[26,27]. All cases within this subgroup harbor a rearrangement of DUX4 to the immunoglobulin heavy chain (IGH) gene or to ERG, leading to DUX4 activation and overexpression. DUX4 encodes a double homeobox transcription factor that is not expressed in normal human B-cell development. Its activation is responsible for ERG deregulation and loss of function. Beside frequent intragenic ERG gene deletion (about 50% of the cases), recurrent alterations in other lymphoid transcription factors are present, including IKZF1 alterations in one third of cases. DUX4/ERG subtype is encountered more often in childhood HR ALL (9.4% vs. 5.2% in SR group) and in adolescents (10.2%); however, its prognosis appears to be excellent, despite the co-existence of IKZF1 deletion[26-29].
ETV6-RUNX1-like ALL
ETV6-RUNX1-like ALL subset regroups B-ALL cases sharing a similar GEP with ETV6-RUNX1-positive ALL but lacking the ETV6-RUNX1 gene fusion. This subgroup represents about 3% of B-ALL and seems to be almost exclusive to childhood ALL[11,27]. They are characterized by frequent co-existing aberrations (gene fusion or copy number alteration) of ETV6 and IKZF1 genes[27,30]. Similar to ETV6-RUNX1 ALL, the ETV6-RUNX1-like phenotype is also associated with a favorable prognosis[27,31].
PAX5-driven subtypes
Aberrations involving the transcription factor PAX5 has recently been individualized as a founding event in B-lymphoid leukemogenesis[11,31]. Two distinct subgroups have been identified within PAX5-driven mutations: PAX5 altered (PAX5alt) ALL, that comprises a diverse spectrum of PAX5 rearrangement, intragenic amplification or sequence mutation, and PAX5 P80R. The latter is characterized by the deleterious PAX5 P80R point mutation coexisting with a near systematic inactivation of the wild-type PAX5 allele either by deletion, loss-of-function mutation or loss of heterozygosity. Frequent signaling pathway mutations arise concomitantly with the PAX5 P80R subtype, mostly involving RAS and JAK-STAT pathways[11]. The PAX5alt subtype predominates in childhood ALL (10%) compared to adult ALL (7%) and is associated with intermediate prognosis[31]. In contrast, PAX5 P80R increases in frequency with age and also confers an intermediate prognosis[11].
MEF2D and ZNF384 rearrangements
The identification of recurrent fusions involving MEF2D and ZNF384 highly suggests their role as oncogenic drivers in B-ALL. Both MEF2D and ZNF384-rearranged (MEF2Dr and ZNF384r) ALL harbor a profile of activated transcription factor gene and disruption in B-cell development, but still present a distinct GEP allowing for the definition of two new subgroups[31,32]. MEFD2r and ZNF384r resemble by their multiple fusion partners, the most common being BCL9 for the former and EP300 for the latter[33,34]. These rare subsets are found in adolescents and young adults (AYA) more often than in younger children, each subset totaling roughly 7% of B-ALL in AYA and 4% in children[32,34]. The scarcity of these subgroups lessens the ability to accurately determine their prognostic significance; however, MEFD2 fusions appear to confer a poor prognosis, while ZNF384 fusions are associated with an intermediate prognosis[31,32]. Interestingly, their molecular signatures are characterized by distinct immunophenotypes. MEF2Dr ALL tends to lack CD10 surface marker while expressing CD38; whereas ZNF384r ALL exhibits lineage plasticity and may be found in approximately half of B/myeloid mixed-phenotype acute leukemia (MPAL) with frequent co-expression of myeloid markers (CD13 and CD33) and lack of CD10 expression[33,34]. Considering the lineage aberrancy in a patient with MPAL, ZNF384 fusion may represent a more reliable diagnostic biomarker rather than relying on the immunophenotype[9,35].
Rare newly defined subgroups
Most recently, two different teams have described novel rare subgroups, totaling up to 23 B-ALL subtypes[11,31]. The rarity of these subsets yields uncertain prognostic interpretation that will require further validation. First, fusions in the chromatin regulator NUTM1, described in about 1% of B-ALL, harbor a unique transcriptional signature and enriched among KMT2A-germline infant ALL cases[36]. Secondly, while IKZF1 alterations can be encountered across different molecular subgroups in B-ALL, the missense mutation IKZF1 N159Y reveals a strikingly distinct molecular signature. Finally, despite recurrent IGH rearrangements to multiple partners are often encountered in B-ALL, the rearrangement of IGH to BCL2, MYC and/or BCL6 defines a new subgroup that is present mostly in adult ALL, accounting for 1% of them[11,31]. Thus, the advent of NGS, in particular with the increasing utilization of whole transcriptome analysis or other clinical RNA-based fusion assays, now enables molecular profiling and classification for approximately 95% of all pediatric B-ALL[9,27]. However, it is important to recognize that the prognostic significance of these rare molecular subgroups is limited by the paucity of cases and should be confirmed in large, prospective, multicenter clinical trials. It is unknown whether the prognostic impact of these gene alterations remains independently adverse in the context of modern MRD-directed treatment strategies.
T-lineage ALL
T-cell ALL (T-ALL) represents 15% of pediatric ALL and up to 25% of adult ALL[2,37]. Its prognosis is historically considered inferior to that of B-ALL and remains a HR determinant in several ALL protocols. However, with contemporary intensive risk-adapted treatment, the 5-year event-free survival (EFS) of T-ALL exceeds 85%, thus approximating that of HR B-ALL[8]. Unlike for B-ALL, the incorporation of molecular classification in T-ALL trials is hindered by the heterogeneity of genetic alterations found in T-ALL[8,37]. Immunophenotyping classification was first used to identify the HR subgroup of early T-cell precursor (ETP) ALL which is characterized by lack of CD1a and CD8, dim CD5, and the expression of aberrant stem-cell or myeloid markers[38,39]. About 12% of pediatric T-ALL is categorized as ETP leukemia and 17% in the highly similar group of near-ETP leukemia (identical phenotype but with an elevated CD5 expression). Compared to non-ETP leukemias, these two subgroups are more likely to be resistant to chemotherapy with high frequency of induction failure and MRD positivity[38,40]. However, when stratified by MRD response, the outcome is similar between the different phenotypes and confirms that the prognostic impact of MRD prevails over the immunophenotype[41]. More recently, high-throughput genome sequencing of a large T-ALL cohort identified 106 putative gene alterations and partitioned into eight distinct molecular subgroups [Figure 2]. The prognostic impact of the sentinel genetic alterations in T-ALL remains uncertain and did not outperform MRD-based risk classification[41,42]. T-ALL molecular profiling is characterized by a large number of biologically relevant genomic alterations with 10 to 20 lesions in each individual leukemia[43]. Some anomalies are highly prevalent and occur in the vast majority of T-ALL. For example, activating NOTCH1 mutations and CDKN2A/CDKN2B deletions are present in more that 50% and in up to 70% of T-ALL, respectively[42,44,45]. Alterations encountered in T-ALL can be subdivided into different signaling pathways: transcriptional regulation, NOTCH1 signaling, cell cycle control, kinase activation, epigenetic regulation, RNA processing, ribosomal function and ubiquitination[10,42,43]. Alterations in transcription factor regulation are nearly universal in T-ALL. T-cell receptor rearrangements, placing oncogenic transcription factor genes under the control of strong T-cell specific enhancers, define major T-ALL subtypes with remarkable transcription factor activation: TLX1, LYL1, LMO2/LYL2, TLX3 and NKX2-1. Deregulation in the transcription factors HOXA, LMO1/LMO2 and TAL1 categorizes three other subclasses[42,45]. Signaling pathway activation was observed in 65% of pediatric T-ALL affecting PI3K-AKT (28%), JAK-STAT (25%) and RAS (14%). PI3K-AKT pathway mutations predominate in the TAL1 subtype, while JAK-STAT and RAS pathway mutations are more common in TLX1, TLX3 and HOXA subgroups. Interestingly, some genetic alterations are shared between B- and T-ALL, including KMT2A rearrangements (10%-15% of T-ALL), ABL1 rearrangements, alterations in cell cycle genes (CDKN2A/B) and epigenetic regulators (CREBBP)[42]. The molecular classification partially correlates with the immunophenotypic subgroups. ETP leukemia is found to be enriched in LMO2/LYL2; near ETP leukemia in TAL1 and TLX3-dysregulated subgroups; whereas non-ETP leukemia is associated with TAL1 and TLX1 deregulation[39,42]. ETP ALL harbors recurrent activating mutations in JAK-STAT and RAS-MAPK signaling pathways; some of these mutations are also observed in acute myeloid leukemia. The gene expression profile of ETP ALL, as well as its immunophenotype and mutational landscape, share similarities with stem-cell and myeloid precursors, suggesting that ETP cells of origin may derive from a multipotent stem cell[39,42,46,47].
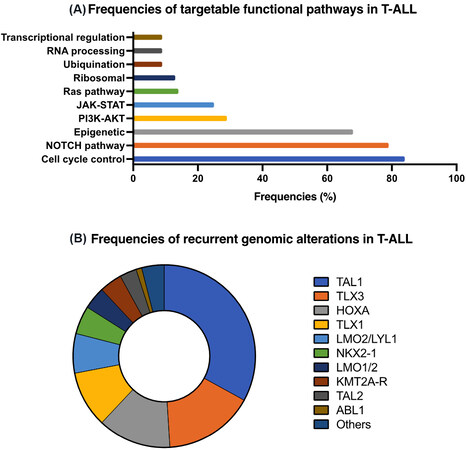
Figure 2. (A) Targetable functional pathways and (B) recurrent genomic alterations in T-lineage acute lymphoblastic leukemia. Frequencies are presented as percentages. T-ALL: T-cell acute lymphoblastic leukemia.
Relapsed ALL
Relapses occur in 10% to 20% of pediatric ALL following first-line therapy and remain one of the leading causes of cancer-related mortality in childhood[10,48]. The genomic landscape of relapsed ALL has been explored by large-scale genome-wide studies involving matched diagnostic-relapsed leukemia samples. The mutational burden at relapse is increased compared to that at diagnosis, with frequent acquisition of genetic alterations that were absent at initial presentation[49,50]. The mutations present at diagnosis and relapse involve several similar pathways: RAS signaling (NRAS, KRAS, PTPN11, FLT3), JAK-STAT pathway (ILR7), NOTCH1 signaling (NOTCH1, FBXW7), transcription factors of lymphoid development (IKZF1, ETV6, PAX5), cell cycle control (CDKN2A/B, TP53) and epigenetic modulators (KMD6A)[49,51]. Some of the mutations retain from diagnosis to relapse, others are volatile and can either be lost or gained at relapse. Volatile dynamics are more likely to be observed with FLT3, JAK2 and RAS pathway mutations, while mutations of IKZF1 and CREBBP are rarely lost at relapse when present at diagnosis[48,52]. Most of the mutations identified at relapse may evolve from a subclonal population. Rarely, secondary leukemias may arise and do not share any genomic signature with the initial leukemia[49,50,52]. However, studies analyzing the clonal lineage with highly sensitive methods for mutation tracking were able to detect the ancestral clone, questioning the real prevalence of secondary leukemias. Even some relapses characterized by a lineage shift reveal mutational similarities between diagnosis and relapse, pointing to a unique clonal origin[35,48,49]. Recently, some mutations exclusive to relapsed ALL samples have been identified (NCOR2, USH2A, NT5C2 and PRPS1), these mutations are never observed at diagnosis and suggest that they are therapy-induced[48,53]. Similarly, a number of mutations enriched at relapse confer resistance to therapeutic agents commonly used in ALL and could have been selected during treatment. These mutations can alter sensitivity to glucocorticoids (NR3C1, NR3C2, CREBBP, WHCS1), purines analogs (NT5C2, PRPS1, PRPS2, MSH2, MSH6, PMS2), methotrexate (FPGS) and anthracycline/vincristine (TP53)[48,51,53-55]. The clonal evolution of these alterations may account for the different relapse patterns. For instance, early relapse arises from a minor subclone that survives and acquires a secondary resistant mutation during treatment, whereas very early relapse emerges from a pre-existing resistant clone[51].
INHERITED GENETIC VARIANTS IN PEDIATRIC ALL
Large scale genome analysis has also unraveled novel germline predisposition syndromes. Before genomic characterization, only a few mendelian diseases were associated with a higher incidence of childhood ALL, such as Down syndrome (DS), ataxia telangiectasia, Bloom syndrome and constitutional mismatch repair deficiency[56-58]. The children with DS have a 10- to 20-fold increased risk of developing ALL compared to the general population[57,59]. DS-ALL, unlike DS-related acute myeloid leukemia, occurs rarely in the first year of life and is associated with an inferior outcome in comparison to non-DS ALL. The lower survival can be attributed to a higher relapse rate and a susceptibility to chemotherapy in DS patients who display a heightened treatment-related toxicity. Nevertheless, the survival difference is not observed across different clinical trials and might be explained by the specific genomic landscape of DS-ALL[57,60]. ALL from DS patients is less likely to carry favorable cytogenetic alterations, such as ETV6-RUNX1 or hyperdiploidy, and is enriched in CRLF2 rearrangements, in 50% to 60% of DS-ALL cases, with frequent JAK2 mutations, a similar profile to what is encountered in Ph-like ALL[59-61]. However, in contrast to HR B-ALL without DS, the presence of CRLF2 alterations in DS-ALL does not correlate with a worse prognosis, as reported by a large retrospective cohort comprising 317 DS-ALL patients treated on COG clinical trials between 2003 and 2016[62].
New germline alterations have been recently discovered since the NGS advent. The germline variants conferring ALL susceptibility can be subdivided into two categories: (1) rare pathogenic germline variants with high penetrance (TP53 and transcription factor genes ETV6, PAX5 and IKZF1) found in families with high ALL incidence; (2) a growing number of common inherited variants with low penetrance detected via genome wide association studies (GWAS) (ARID5B, CEBPE, CDKN2A/B, GATA3, PIP4K2A, TP63)[56,63-65]. Similar to somatic alterations in IKZF1, germline mutation alters IKAROS function and affects treatment sensitivity both in vivo and in vitro by reducing dexamethasone and dasatinib tyrosine kinase activity in leukemic cells[66]. Some of these inherited variants predispose to specific ALL molecular subgroups. Inherited TP53 mutations, causing Li-Fraumeni syndrome, occur in about 50% of low-hypodiploid ALL[64]. Variants in GATA3 are strongly associated with Ph-like ALL, while TP63 variants preferably relate to the ETV6-RUNX1 subgroup and PIP4K2A to the hyperdiploid ALL[65,67-69]. Finally, GWAS has uncovered the association of inherited gene polymorphisms and drug sensitivity or toxicity and laid the foundation for clinical pharmacogenomics. The most significant are TPMT and NUDT15 variants responsible for thiopurine-induced myelosuppression[65]. The prevalence of these variants follows ethnic variations; the former being more frequent in African descendent and the latter in people from East Asian or Hispanic origins. Heterozygous patients for these variants tolerate lower dose of thiopurine, approximately 60% to 70% of the usual dosing, while homozygous patients exhibit extreme mercaptopurine sensitivity and necessitate drastic dose reduction to 10% of the intended dose. Pre-emptive screening for these variants is now recommended for pharmacogenomics-based therapy adjustment[70].
TARGETED PRECISION MEDICINE OPPORTUNITIES
The most spectacular survival improvement for relapsed and refractory (R/R) ALL has been achieved with the recent advent of immunotherapy. Bispecific T-cell engager antibody (e.g., blinatumomab), antibody-drug conjugates (e.g., inotuzumab ozogamicin), or cellular therapy (e.g., chimeric antigen receptor T-cells), have shown impressive response in heavily pretreated R/R ALL[71-73]. Immunotherapy-based treatment strategies, targeting common ALL surface antigens, have the advantage of being agnostic to sentinel genetic alterations, and therefore, can be applied to a broader patient population[10]. Thus, there is still room for improvement since a non-negligible proportion of patients fail to respond or relapse after immunotherapy. We hereby review emergent molecularly targeted therapies and precision medicine opportunities in pediatric ALL with the potential for further survival improvement.
Ph+ ALL
The success of tyrosine kinase inhibitor (TKI)-based treatment in Ph+ ALL is certainly one of the most eloquent examples of precision oncology. Until the early 21st century, Ph+ ALL, which concerns 3% to 5% of pediatric ALL, had a dismal outcome. Despite high-intensity chemotherapy followed by allogenic hematopoietic stem cell transplantation (HSCT) intensification in first complete remission, less than 50% of the patients were long-term survivors[74]. Following the remarkable clinical efficacy of the BCR-ABL TKI, imatinib, in chronic myeloid leukemia, TKI became a therapeutic opportunity for Ph+ ALL[8]. Clinical trials from the COG and the European EsPhALL consortia have demonstrated a robust survival benefit in Ph+ ALL with the incorporation of imatinib combined to an intensive chemotherapy backbone, thus challenging the indication of HSCT in first remission in the TKI era. With imatinib-based regimens, almost 70% of Ph+ ALL patients can avoid HSCT and achieve durable remissions[75-78]. The second generation BCR-ABL1 TKI, dasatinib, is an alternative to imatinib in combination to intensive chemotherapy that has been tested in COG and EsPhALL non-randomized clinical trials. In these studies, dasatinib did not contribute to further improve the clinical outcome, showing a similar efficacy to historical imatinib-based regimens[79,80]. However, earlier introduction of protracted TKI administration by mid-induction appears to increase the achievement of post-induction MRD negativity, then reducing the need of HSCT consolidation in first remission. The Chinese Children’s Cancer Group (CCCG) ALL-2015 phase 3 randomized trial compared the combination of either dasatinib or imatinib to the St. Jude Total Therapy chemotherapy backbone and suggested dasatinib’s superiority over imatinib with regards to EFS and OS. This result needs to be confirmed as the median follow-up remains relatively short. In addition, the outcomes in the imatinib arm were unexpectedly inferior compared to those of prior imatinib-based regimens in COG or EsPhALL trials and the TKI dosage used was different than prior studies[81]. The promising success of TKI and chemotherapy combination in the treatment of Ph+ ALL has established a new treatment paradigm for ALL in the molecular era, nourishing the hope of new therapeutic opportunities by targeting novel oncogenic drivers in ALL. Recently, the combination of TKI and immunotherapy further expands the precision medicine paradigm in Ph+ ALL. The chemotherapy-free phase 2 D-ALBA study demonstrated early promising results in adults with newly-diagnosed Ph+ ALL. The treatment consists of 85-day induction phase combining dasatinib and glucocorticoids followed by a consolidation with 2 cycles or more of the CD3/CD19 bi-specific antibody, blinatumomab, in association with dasatinib. Interestingly, after induction therapy with dasatinib and glucocorticoids only, 98% of the patients achieved a complete response, and 29% a molecular response. After 2 cycles of blinatumomab and dasatinib consolidation, the molecular response rate exceeded 60%[82].
Ph-like ALL
The clinical and biologic similarities between Ph-like ALL and Ph+ ALL provide the rationale to model tailored TKI-based therapy in Ph-like ALL with an anticipated efficacy similar to that observed in Ph+ ALL. In vitro and in vivo evidence have reinforced the putative sensitivity of Ph-like ALL to selected TKIs by demonstrating activity of ABL inhibitors, imatinib or dasatinib, and JAK inhibitor, ruxolitinib, for those harboring ABL-class fusions and JAK-STAT pathway mutations, respectively[19,20,83-85]. There is a growing body of clinical evidence from case reports or small patient series to confirm the preclinical efficacy of ABL and JAK inhibitors in Ph-like ALL[86-88]. Preclinical and clinical reports have also demonstrated the sensitivity of NTRK inhibitor, larotrectinib, or ALK inhibitor, crizotinib, for the rare ETV6-NTRK3 Ph-like ALL[19,89,90]. Recently, mutations conferring resistance to imatinib and dasatinib have been identified in relapsed EBF1-PDGFRB Ph-like ALL, raising the necessity to monitor kinase domain mutations over the course of therapy to guide TKI selection[91]. Thanks to the progress in genomic characterization of ALL, Ph-like ALL has become a new paradigm of tailored precision medicine, but prospective clinical trials are much needed to confirm the benefit of TKI and chemotherapy in Ph-like ALL. Several ongoing clinical trials should shortly answer some of these urgent questions [Total XVII (NCT03117751); AALL1131 (NCT01406756); AALL1521 (NCT02723994)] [Table 1].
Precision medicine opportunities in pediatric acute lymphoblastic leukemia
| ALL subtypes | Therapies | Preclinical - single patient experience | Clinical trials | Phase |
| Ph+ ALL | Imatinib | AALL0031 (NCT00022737) EsPhALL2010 (NCT00287105) CCCG-ALL-2015 EsPhALL 2017/COG AALL1631 (NCT03007147) | 3 2 3 3 | |
| Dasatinib | AALL0622 (NCT00720109) AALL1122 (NCT01460160) CCCG-ALL-2015 | 2 2 3 | ||
| Ponatinib | NCT04501614 | 1/2 | ||
| Rexinoids[92] | ||||
| FAK inhibitors[93] | ||||
| Ph-like ALL - With ABL-class fusions | Dasatinib | AALL1131 (NCT02883049) Total Therapy XVII (NCT03117751) DFCI ALL 16-001 (NCT03020030) | 3 3 3 | |
| - With JAK-STAT pathway lesions | Ruxolitinib | AALL1521 (NCT02723994) Total Therapy XVII (NCT03117751) | 2 3 | |
| - With NTRK fusions | Larotrectinib | ADVL1823 (NCT03834961) | 2 | |
| PI3K/AKT/mTOR inhibitors[85] | ||||
| KMT2A-R ALL | Lestaurtinib | AALL0631 (NCT00557193) | 3 | |
| Azacitidine | AALL15P1 (NCT02828358) | 2 | ||
| Vorinostat/ Bortezomib | TINI (NCT02553460) | 1/2 | ||
| Menin inhibitors | NCT04811560 (adults only) | 1 | ||
| DOT1L inhibitors | NCT02141828 | 1 | ||
| Venetoclax | NCT03826992 | 1 | ||
| Hypodiploid ALL | Venetoclax[94] | |||
| ZNF384-R ALL | FLT3 inhibitors[95] | |||
| MEF2D-R ALL | HDAC inhibitors[33] | |||
| T-ALL | Venetoclax | NCT00501826 | 2 | |
| - NOTCH pathway mutations | Y-secretase inhibitors Soluble notch proteins Mastermind inhibiting peptides[96] | |||
| - JAK-STAT mutations - ETP ALL | Ruxolitinib | Total Therapy XVII (NCT03117751) | 3 | |
| - PI3K/AKT/mTOR pathway mutations | Everolimus | PI3K/AKT/mTOR inhibitors[97] | DFCI 11-237 (NCT01523977) | 1 |
| Farnesyltransferase inhibitors[98] | ||||
| - Ubiquination | Bortezomib Carfilzomib | AALL1231 (NCT02112916) Total Therapy XVII (NCT03117751) CFZ008 (NCT02303821) | 3 3 1 | |
| B and T-ALL - Cell cycle control | Palbociclib Ribociclib | AINV18P1 (NCT03792256) NCT03740334 | 1 1 | |
| - Ras pathway mutations | Selumetinib | NCT03705507 | 1/2 |
Proapoptotic targeted therapy
Proapoptotic agents, acting as cell death inducers by overcoming chemoresistance, represent another promising therapeutic opportunity for ALL[99]. KMT2A rearrangements, frequently observed in infant ALL, are known to confer resistance to apoptosis, emphasizing the interest of apoptotic inducer for this subgroup[100,101]. A study on xenografts confirmed the activity of the selective BCL-2 inhibitor, venetoclax, for KMT2Ar ALL models, while other ALL subtypes escaped venetoclax inhibition by activating the bcl-x pathway[99]. More recently, venetoclax appeared to provide in vitro and in vivo efficacy for controlling the leukemic burden in hypodiploid ALL, in TCF3-HLF ALL and in ETP ALL[94,102,103]. The potential activity in T-ALL has been reinforced by case reports of venetoclax activity, in association with decitabine or bortezomib[104,105]. A pediatric phase 1 clinical trial of venetoclax with chemotherapy combination showed good tolerance to the regimen and encouraging clinical activity in ALL patients[106]. The addition of the BCL-2/BCL-XL/BCL-W, navitoclax, to venetoclax and chemotherapy seems to enhance the clinical activity of venetoclax by overcoming escape pathways. A phase 1 study with this combination reported a promising activity of 86% of response in pediatric relapsed/refractory ALL patients, among whom 56% achieved negative MRD[107]. Targeting proapoptotic pathway offers an optimistic outlook in the landscape of R/R ALL treatment, but the best combination and ideal target population have yet to be determined.
Therapeutic opportunities for rare molecular subgroups
The modern genomic taxonomy of ALL identified some therapeutic susceptibility in rare molecular subsets, but clinical validation is required. The extensive genomic characterization by whole genome and transcriptome analysis of a ZNF384-rearranged ALL patient revealed a highly aberrant FLT3 overexpression. This patient, presenting with refractory disease, happened to be highly sensitive to the FLT3 inhibitor, sunitinib, leading to deep MRD-negative response and long-term survival[95]. This case illustrates how the combination of genome and transcriptome analysis can lead to the identification of unsuspected therapeutic vulnerabilities. Another example is the constant overexpression of the histone deacetylase HDAC9, a transcriptional target of MEF2D, observed in MEF2D-rearranged ALL. Xenograft models were used to test the therapeutic vulnerability of MEF2Dr ALL to the HDAC inhibitor, panabinostat, and showed exquisite in vivo sensitivity[33]. No clinical experience has been reported yet. Finally, IKZF1 inactivating mutations induce in vitro stem cell and adhesive properties and alter the response to dasatinib in BCR-ABL1 mouse models. Retinoids and focal adhesion kinase (FAK) inhibitors have the ability to reverse these mechanisms, thus restoring dasatinib sensitivity[92,93]. Retinoids and FAK inhibitors constitute potential therapeutic avenues that can be explored for IKZF1-mutated Ph+ ALL and Ph-like ALL, and for the IKZF1 N159Y subgroup.
A major challenge for the translation and implementation of the modern ALL taxonomy into pragmatic clinical algorithms is the limited access to comprehensive NGS platforms and their relatively long turnaround time, making it challenging for real time patient’s care. Nevertheless, a number of clinically-validated NGS assays are increasingly available and being incorporated into frontline ALL trials. For example, the Rapid Heme Panel, a DNA-based NGS diagnostic assay currently used in the DFCI ALL 16-001 study for the detection of sequence mutations and CNAs, is able to deliver results within 10 days for risk stratification[108]. Gene expression profiling for the identification of Ph-like ALL has now been largely replaced by the TaqMan low-density array (LDA) microfluidic card measuring the expression of 8- or 15-gene panels to determine the Ph-like ALL signature. This LDA-based approach has provided a rapid and cost-effective screening modality to identify patients with probable Ph-like ALL (LDA-positive) who require further detailed genomic characterization. To identify targetable kinase-activating alterations, the ArcherDx FusionPlex Heme panel uses anchored multiplex PCR-based enrichment with the ability to detect novel fusions involving 87 genes associated with hematologic malignancies[109]. The AIEOP-BFM consortium is now incorporating array comparative genomic hybridization to identify the IKZF1plus profile and panel-based RNA sequencing to detect targetable gene fusions for upfront risk stratification in their frontline ALL trial[110]. At the level of a single center, the St. Jude Children’s Research Hospital attests that a full DNA- and RNA-based sequencing approach is feasible within 4 weeks and suitable for integration into patient’s real-time management[111]. The modernization of sequencing technologies and the development of standardized bioinformatic pipelines for timely data analysis should facilitate routine clinical implementation of genomic profiling for pediatric ALL.
CONCLUSION
Advances of high-throughput sequencing technologies redefine the genomic portrait of ALL and modernize ALL classification, with only a marginal proportion of patients remaining unclassified in the current molecular landscape. Beside Ph+ and Ph-like ALL for whom robust preclinical and clinical evidence supports the prospective assessment of TKI and chemotherapy in frontline trials, a substantial proportion of molecular subsets, particularly those associated with unfavorable outcomes, still lacks access to key therapeutic targets and relevant precision medicine trials. Continuous efforts in elucidating the functional mechanisms underlying the subtype-defining alterations in ALL is essential to expand the spectrum of novel targeted therapies. Nevertheless, the development of new therapeutic options and consequently the design of clinical trials are hindered by the growing number of molecular subsets, each accounting for a small proportion of ALL. Large international clinical trials are therefore critical to explore innovative treatments combining chemo-, immuno- and targeted therapies, with the objectives of improving survival in HR subtypes and reducing toxicity in low-risk ALL. The current molecular era of ALL present unique challenges but also offers exciting opportunities for paradigm-shifting therapies.
DECLARATIONS
Authors’ contributionsPerformed the literature review and wrote the manuscript: Santiago R, Tran TH
Both authors contributed equally to this work.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 2012;30:1663-9.
2. Pui CH, Yang JJ, Hunger SP, et al. childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol 2015;33:2938-48.
3. Santiago R, Vairy S, Sinnett D, Krajinovic M, Bittencourt H. Novel therapy for childhood acute lymphoblastic leukemia. Expert Opin Pharmacother 2017;18:1081-99.
4. Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol 1996;14:18-24.
5. Harrison CJ, Moorman AV, Schwab C, Iacobucci I, Mullighan C. . Cytogenetics and molecular genetics. In: Vora A, editor. Childhood acute lymphoblastic leukemia. Cham: Springer International Publishing; 2017. p. 61-98.
6. Schultz KR, Pullen DJ, Sather HN, et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood 2007;109:926-35.
7. Vrooman LM, Blonquist TM, Harris MH, et al. Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv 2018;2:1449-58.
8. Tasian SK, Hunger SP. Genomic characterization of paediatric acute lymphoblastic leukaemia: an opportunity for precision medicine therapeutics. Br J Haematol 2017;176:867-82.
9. Mullighan CG. How advanced are we in targeting novel subtypes of ALL? Best Pract Res Clin Haematol 2019;32:101095.
10. Tran TH, Hunger SP. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin Cancer Biol 2020:S1044-579X(20)30218.
11. Gu Z, Churchman ML, Roberts KG, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 2019;51:296-307.
12. Yeoh E, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002;1:133-43.
13. Kang H, Chen IM, Wilson CS, et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood 2010;115:1394-405.
14. Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 2010;116:4874-84.
15. Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009;10:125-34.
16. Mullighan CG, Su X, Zhang J, et al. Children's Oncology Group. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009;360:470-80.
17. Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010;115:5312-21.
18. Loh ML, Zhang J, Harvey RC, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood 2013;121:485-8.
19. Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014;371:1005-15.
20. Tran TH, Loh ML. Ph-like acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2016;2016:561-6.
21. Iacobucci I, Storlazzi CT, Cilloni D, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell'Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood 2009;114:2159-67.
22. Vairy S, Tran TH. IKZF1 alterations in acute lymphoblastic leukemia: The good, the bad and the ugly. Blood Rev 2020;44:100677.
23. Tran TH, Harris MH, Nguyen JV, et al. Prognostic impact of kinase-activating fusions and IKZF1 deletions in pediatric high-risk B-lineage acute lymphoblastic leukemia. Blood Adv 2018;2:529-33.
24. Chen IM, Harvey RC, Mullighan CG, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 2012;119:3512-22.
25. Stanulla M, Dagdan E, Zaliova M, et al. TRANSCALL Consortium. IKZF1plus defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol 2018;36:1240-9.
26. Zhang J, McCastlain K, Yoshihara H, et al. St. Jude Children's Research Hospital-Washington University Pediatric Cancer Genome Project. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 2016;48:1481-9.
27. Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 2016;7:11790.
28. Clappier E, Auclerc MF, Rapion J, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 2014;28:70-7.
29. Zaliova M, Zimmermannova O, Dörge P, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 2014;28:182-5.
30. Zaliova M, Kotrova M, Bresolin S, et al. ETV6/RUNX1-like acute lymphoblastic leukemia: A novel B-cell precursor leukemia subtype associated with the CD27/CD44 immunophenotype. Genes Chromosomes Cancer 2017;56:608-16.
31. Li JF, Dai YT, Lilljebjörn H, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A 2018;115:E11711-20.
32. Liu YF, Wang BY, Zhang WN, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine 2016;8:173-83.
33. Gu Z, Churchman M, Roberts K, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 2016;7:13331.
34. Hirabayashi S, Ohki K, Nakabayashi K, et al. Tokyo Children’s Cancer Study Group (TCCSG). ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017;102:118-29.
35. Alexander TB, Gu Z, Iacobucci I, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018;562:373-9.
36. Fazio G, Bardini M, De Lorenzo P, et al. Recurrent genetic fusions redefine MLL germ line acute lymphoblastic leukemia in infants. Blood 2021;137:1980-4.
37. Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol 2008;8:380-90.
38. Coustan-smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 2009;10:147-56.
39. Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012;481:157-63.
40. Patrick K, Wade R, Goulden N, et al. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol 2014;166:421-4.
41. Wood BL, Winter SS, Dunsmore KP, et al. T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in Children’s Oncology Group (COG) Study AALL0434. Blood 2014;124:1.
42. Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 2017;49:1211-8.
43. Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood 2017;129:1113-23.
44. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004;306:269-71.
45. Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest 2012;122:3398-406.
46. Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 2015;125:1759-67.
47. Neumann M, Coskun E, Fransecky L, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PLoS One 2013;8:e53190.
48. Waanders E, Gu Z, Dobson SM, et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov 2020;1:96-111.
49. Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 2015;6:6604.
50. Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008;322:1377-80.
51. Li B, Brady SW, Ma X, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020;135:41-55.
52. Schroeder MP, Bastian L, Eckert C, et al. Integrated analysis of relapsed B-cell precursor Acute Lymphoblastic Leukemia identifies subtype-specific cytokine and metabolic signatures. Sci Rep 2019;9:4188.
53. Li B, Li H, Bai Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med 2015;21:563-71.
54. Meyer JA, Wang J, Hogan LE, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 2013;45:290-4.
55. Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011;471:235-9.
56. Treviño LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 2009;41:1001-5.
58. Ripperger T, Schlegelberger B. Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur J Med Genet 2016;59:133-42.
59. Brown AL, de Smith AJ, Gant VU, et al. Inherited genetic susceptibility to acute lymphoblastic leukemia in Down syndrome. Blood 2019;134:1227-37.
60. Maloney KW, Carroll WL, Carroll AJ, et al. Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 2010;116:1045-50.
61. Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 2009;41:1243-6.
62. Rabin KR, Devidas M, Chen Z, et al. Outcomes of patients with Down syndrome and CRLF2 -overexpressing acute lymphoblastic leukemia (ALL): a report from the Children's Oncology Group (COG). Blood 2020;136:44-5.
63. Gocho Y, Yang JJ. Genetic defects in hematopoietic transcription factors and predisposition to acute lymphoblastic leukemia. Blood 2019;134:793-7.
64. Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013;45:242-52.
65. Moriyama T, Relling MV, Yang JJ. Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood 2015;125:3988-95.
66. Churchman ML, Qian M, Te Kronnie G, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell 2018;33:937-48.e8.
67. Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet 2013;45:1494-8.
68. Walsh KM, de Smith AJ, Chokkalingam AP, et al. Novel childhood ALL susceptibility locus BMI1-PIP4K2A is specifically associated with the hyperdiploid subtype. Blood 2013;121:4808-9.
69. Ellinghaus E, Stanulla M, Richter G, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 2012;26:902-9.
70. Yang JJ, Landier W, Yang W, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 2015;33:1235-42.
71. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;378:439-48.
72. O'brien MM, Ji L, Shah NN, et al. A Phase 2 Trial of inotuzumab ozogamicin (InO) in children and young adults with relapsed or refractory (R/R) CD22+ B-acute lymphoblastic leukemia (B-ALL): results from Children's Oncology Group Protocol AALL1621. Blood 2019;134:741.
73. Brown PA, Ji L, Xu X, et al. Effect of postreinduction therapy consolidation with blinatumomab vs chemotherapy on disease-free survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA 2021;325:833-42.
74. Aricò M, Schrappe M, Hunger SP, et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 2010;28:4755-61.
75. Biondi A, Gandemer V, De Lorenzo P, et al. Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): a prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol 2018;5:e641-52.
76. Schultz KR, Carroll A, Heerema NA, et al. Children’s Oncology Group. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 2014;28:1467-71.
77. Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 2009;27:5175-81.
78. Biondi A, Schrappe M, De Lorenzo P, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 2012;13:936-45.
79. Hunger SP, Saha V, Devidas M, et al. CA180-372: An International Collaborative Phase 2 Trial of dasatinib and chemotherapy in pediatric patients with newly diagnosed philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL). Blood 2017;130:98-98.
80. Slayton WB, Schultz KR, Kairalla JA, et al. Dasatinib plus intensive chemotherapy in children, adolescents, and young adults with philadelphia chromosome-positive acute lymphoblastic leukemia: results of Children's Oncology Group Trial AALL0622. J Clin Oncol 2018;36:2306-14.
81. Shen S, Chen X, Cai J, et al. Effect of dasatinib vs imatinib in the treatment of pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: a randomized clinical trial. JAMA Oncol 2020;6:358-66.
82. Foà R, Bassan R, Vitale A, et al. GIMEMA Investigators. Dasatinib-blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N Engl J Med 2020;383:1613-23.
83. Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 2012;22:153-66.
84. Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2012;120:3510-8.
85. Tasian SK, Doral MY, Borowitz MJ, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood 2012;120:833-42.
86. Tanasi I, Ba I, Sirvent N, et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood 2019;134:1351-5.
87. Ding YY, Stern JW, Jubelirer TF, et al. Clinical efficacy of ruxolitinib and chemotherapy in a child with Philadelphia chromosome-like acute lymphoblastic leukemia with GOLGA5-JAK2 fusion and induction failure. Haematologica 2018;103:e427-31.
88. Cario G, Leoni V, Conter V, et al. Relapses and treatment-related events contributed equally to poor prognosis in children with ABL-class fusion positive B-cell acute lymphoblastic leukemia treated according to AIEOP-BFM protocols. Haematologica 2020;105:1887-94.
89. Roberts KG, Janke LJ, Zhao Y, et al. ETV6-NTRK3 induces aggressive acute lymphoblastic leukemia highly sensitive to selective TRK inhibition. Blood 2018;132:861-5.
90. Nardi V, Ku N, Frigault MJ, et al. Clinical response to larotrectinib in adult Philadelphia chromosome-like ALL with cryptic ETV6-NTRK3 rearrangement. Blood Adv 2020;4:106-11.
91. Tran TH, Nguyen JV, Stecula A, et al. The EBF1-PDGFRB T681I mutation is highly resistant to imatinib and dasatinib in vitro and detectable in clinical samples prior to treatment. Haematologica 2021; doi: 10.3324/haematol.2020.261354.
92. Churchman ML, Low J, Qu C, et al. Efficacy of retinoids in IKZF1-mutated BCR-ABL1 acute lymphoblastic leukemia. Cancer Cell 2015;28:343-56.
93. Churchman ML, Evans K, Richmond J, et al. Synergism of FAK and tyrosine kinase inhibition in Ph+ B-ALL. JCI Insight 2016;1:86082.
94. Diaz-Flores E, Comeaux EQ, Kim KL, et al. Bcl-2 is a therapeutic target for hypodiploid B-lineage acute lymphoblastic leukemia. Cancer Res 2019;79:2339-51.
95. Griffith M, Griffith OL, Krysiak K, et al. Comprehensive genomic analysis reveals FLT3 activation and a therapeutic strategy for a patient with relapsed adult B-lymphoblastic leukemia. Exp Hematol 2016;44:603-13.
96. Habets RA, de Bock CE, Serneels L, et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci Transl Med 2019;11:eaau6246.
97. Yuan T, Yang Y, Chen J, et al. Regulation of PI3K signaling in T-cell acute lymphoblastic leukemia: a novel PTEN/Ikaros/miR-26b mechanism reveals a critical targetable role for PIK3CD. Leukemia 2017;31:2355-64.
98. Alonso-Alonso R, Mondéjar R, Martínez N, et al. Identification of tipifarnib sensitivity biomarkers in T-cell acute lymphoblastic leukemia and T-cell lymphoma. Sci Rep 2020;10:6721.
99. Khaw SL, Suryani S, Evans K, et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood 2016;128:1382-95.
100. Gaussmann A, Wenger T, Eberle I, et al. Combined effects of the two reciprocal t(4;11) fusion proteins MLL.AF4 and AF4.MLL confer resistance to apoptosis, cell cycling capacity and growth transformation. Oncogene 2007;26:3352-63.
101. Inukai T, Zhang X, Goto M, et al. Resistance of infant leukemia with MLL rearrangement to tumor necrosis factor-related apoptosis-inducing ligand: a possible mechanism for poor sensitivity to antitumor immunity. Leukemia 2006;20:2119-29.
102. Peirs S, Matthijssens F, Goossens S, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood 2014;124:3738-47.
103. Fischer U, Forster M, Rinaldi A, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 2015;47:1020-9.
104. Farhadfar N, Li Y, May WS, Adams CB. Venetoclax and decitabine for treatment of relapsed T-cell acute lymphoblastic leukemia: a case report and review of literature. Hematol Oncol Stem Cell Ther 2021;14:246-51.
105. La Starza R, Cambò B, Pierini A, et al. Venetoclax and bortezomib in relapsed/refractory early T-cell precursor acute lymphoblastic leukemia. JCO Precis Oncol 2019;3:PO.
106. Karol SE, Cooper TM, Bittencourt H, et al. Safety, efficacy, and PK of the BCL2 inhibitor venetoclax in combination with chemotherapy in pediatric and young adult patients with relapsed/refractory acute myeloid leukemia and acute lymphoblastic leukemia: Phase 1 Study. Blood 2019;134:2649.
107. Lacayo NJ, Pullarkat VA, Stock W, et al. Safety and efficacy of venetoclax in combination with navitoclax in adult and pediatric relapsed/refractory acute lymphoblastic leukemia and lymphoblastic lymphoma. Blood 2019;134:285.
108. Kluk MJ, Lindsley RC, Aster JC, et al. Validation and implementation of a custom next-generation sequencing clinical assay for hematologic malignancies. J Mol Diagn 2016;18:507-15.
109. Harvey RC, Tasian SK. Clinical diagnostics and treatment strategies for Philadelphia chromosome-like acute lymphoblastic leukemia. Blood Adv 2020;4:218-28.
110. Schieck M, Lentes J, Thomay K, et al. Implementation of RNA sequencing and array CGH in the diagnostic workflow of the AIEOP-BFM ALL 2017 trial on acute lymphoblastic leukemia. Ann Hematol 2020;99:809-18.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Santiago R, Tran TH. Genomics and precision medicine in pediatric acute lymphoblastic leukemia. J Transl Genet Genom 2021;5:380-95. http://dx.doi.org/10.20517/jtgg.2021.16
AMA Style
Santiago R, Tran TH. Genomics and precision medicine in pediatric acute lymphoblastic leukemia. Journal of Translational Genetics and Genomics. 2021; 5(4): 380-95. http://dx.doi.org/10.20517/jtgg.2021.16
Chicago/Turabian Style
Santiago, Raoul, Thai Hoa Tran. 2021. "Genomics and precision medicine in pediatric acute lymphoblastic leukemia" Journal of Translational Genetics and Genomics. 5, no.4: 380-95. http://dx.doi.org/10.20517/jtgg.2021.16
ACS Style
Santiago, R.; Tran TH. Genomics and precision medicine in pediatric acute lymphoblastic leukemia. J. Transl. Genet. Genom. 2021, 5, 380-95. http://dx.doi.org/10.20517/jtgg.2021.16
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 25 clicks
Cite This Article 25 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.