
Shared genomic segment analysis in a large high-risk chronic lymphocytic leukemia pedigree implicates CXCR4 in inherited risk
 0
0Abstract
Aim: Chronic lymphocytic leukemia (CLL) has been shown to cluster in families. First-degree relatives of individuals with CLL have an ~8 fold increased risk of developing the malignancy. Strong heritability suggests pedigree studies will have good power to localize pathogenic genes. However, CLL is relatively rare and heterogeneous, complicating ascertainment and analyses. Our goal was to identify CLL risk loci using unique resources available in Utah and methods to address intra-familial heterogeneity.
Methods: We identified a six-generation high-risk CLL pedigree using the Utah Population Database. This pedigree contains 24 CLL cases connected by a common ancestor. We ascertained and genotyped eight CLL cases using a high-density SNP array, and then performed shared genomic segment (SGS) analysis - a method designed for extended high-risk pedigrees that accounts for heterogeneity.
Results: We identified a genome-wide significant region (P = 1.9 × 10-7, LOD-equivalent 5.6) at 2q22.1. The 0.9 Mb region was inherited through 26 meioses and shared by seven of the eight genotyped cases. It sits within a ~6.25 Mb locus identified in a previous linkage study of 206 small CLL families. Our narrow region intersects two genes, including CXCR4 which is highly expressed in CLL cells and implicated in maintenance and progression.
Conclusion: SGS analysis of an extended high-risk CLL pedigree identified the most significant evidence to-date for a 0.9 Mb CLL disease locus at 2q22.1, harboring CXCR4. This discovery contributes to a growing literature implicating CXCR4 in inherited risk to CLL. Investigation of the segregating haplotype in the pedigree will be valuable for elucidating risk variant(s).
Keywords
INTRODUCTION
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia diagnosed in individuals of European ancestry in the United States (5.0/100,000)[1]. CLL has a strong heritable component, and first-degree relatives have a 7.5-8.5 fold elevated risk of developing CLL[2-4]. Therefore, a family-based design is relevant to consider for CLL. However, because CLL is relatively rare, this design presents a challenge in ascertainment of multi-case families. Translational relevance for successful family discoveries includes genetic counselling for at-risk family members and new avenues for understanding biological mechanism towards improved prevention and treatment.
An established family-based statistical approach is linkage analysis. Recombination events are estimated in families which localize regions, and risk haplotypes, which are inherited to affect family members. These haplotypes can subsequently be interrogated to identify specific variants involved in disease pathogenesis. This design boosts power for rarer risk alleles which are enriched in the family setting. In the study of familial CLL, five genome-wide linkage studies have been performed thus far[5-9]. Of these, only one locus has been proposed with genome-wide significance. Linkage analyses in 206 CLL families identified a significant peak at chromosome 2q21.2 (LOD-equivalent 3.11, P = 7.7 × 10-5), and a 1-LOD support interval defining a ~6.25 Mb locus at 2q21.2-2q22.1[7]. The locus contains the gene CXCR4 (C-X-C chemokine receptor type 4) (at 2q22.1), of particular interest due to its key role in B cell lymphopoiesis and maintenance of immature B cells in the bone marrow.
Two family-based whole exome sequencing (WES) studies have been performed[10,11]. Rigorous statistical thresholds that account for the multiple phases and family-based expectations have not yet been defined for direct-to-sequencing family studies, hence these studies remain largely observational in nature. The prior studies focused on finding recurrent rare alleles in families, but the segregation of those alleles was not formally assessed (i.e., how probabilities for the findings are influenced by allele frequency, non-sharing in cases, and unaffected carriers). In a study of 59 small families, Goldin et al.[10], focused on identifying coding variants recurrent within and shared across at least two families. They identified 6 families recurrent for an allele in ITGB2 [rs2230531 at 21q22.3, minor allele frequency (MAF) = 0.007]. However, recurrence of this variant was not replicated in a follow-up study of ITGB2 in 47 small families[12]. In a WES study of 66 families (no restriction made to sharing across multiple families), ITGB2 variants were not identified, but four different coding variants were found to be recurrent within 7 different small families, all in genes involving the shelterin complex (four in POT1, one in ACD and two TERF2IP at 7q31.33, 16q22.1 and 16q23.1, respectively)[11].
One family study used genome-wide genotype data to identify germline copy number variants (CNV) in CLL families occurring at regions known to be commonly aberrant in malignant CLL cells. This identified two germline CNVs: a mutation at 13q involving DLEU7 and a gain at 6p including IRF4. Each was shared by a single CLL sib-pair[13]. These findings have yet to be replicated.
The scarcity of CLL family resources, heterogeneity across families and the likely complexity of the disease mechanism (multiple genes, multiple alleles, incomplete penetrance, and sporadic cases) leads to challenges in uncovering inheritable genetic abnormalities. Our goal was to identify CLL risk loci using unique resources available in Utah through the Utah Population Database (UPDB) to identify large, extended, high-risk pedigrees and a powerful new method specifically designed for large pedigrees and to address heterogeneity.
The UPDB includes a 16-generation genealogy of approximately 5 million people with at least one event in Utah that is record-linked to statewide cancer records since 1966 from the NCI Surveillance, Epidemiology, and End Results (SEER) Program Utah Cancer Registry (UCR) and state vital records[14]. Within the UPDB, ancestors whose descendants have an increased incidence of malignances as compared to internal cancer rate controls and years at risk can be identified and studied as high-risk pedigrees.
Shared genomic segment (SGS) analysis is a recombinant-based family analysis (“linkage-like”), developed to identify regions that segregate to cases in an extended high-risk pedigree[15]. When available to study, a single large pedigree can increase homogeneity, garner equivalent power to many small pedigrees, and be sufficient alone to declare genome-wide significance. However, full likelihood-based linkage approaches are intractable in very large pedigrees. Furthermore, traditional linkage methods are not robust to substantial intra-familial heterogeneity (sporadic cases), which must be accounted for in very large pedigrees. To combat this, SGS identifies long stretches of consecutive identity-by-state (IBS) alleles to infer shared inherited identity-by-descent (IBD) haplotypes. The algorithm iterates over (and corrects for) assessment of subsets of cases to account for possible sporadic cases. Overall, SGS is the ideal method for investigating disease risk loci shared by a common founder in large pedigrees.
Here, we use the UPDB to identify a six-generation high-risk CLL pedigree, the largest CLL family studied to-date. We performed SGS to identify inherited risk loci likely to harbor disease genes for CLL.
METHODS
Identification and ascertainment of the high-risk pedigree
The UPDB was used to identify ancestors whose descendants showed a statistical excess of CLL (P < 0.05). Expectation was based on internal disease rates based on birth cohort, sex, birth place (in/outside Utah) and years at risk. These were considered high-risk CLL pedigrees. Once identified, living CLL cases within high-risk pedigrees were made aware of the study by representatives of the UCR, and those interested were invited to participate. Cases and family members wishing to be part of the study were subsequently enrolled by the study team, including informed consent, questionnaires and biospecimens. Individuals in 23 high-risk CLL pedigrees were enrolled as previously described[16]. Only one six-generation pedigree with 24 CLL cases contained sufficient meioses (m ≥ 15) between sampled CLL cases for SGS analysis[17]. Figure 1A illustrates all 24 cases in the pedigree. Figure 1B shows the reduced structure containing only the eight sampled CLL cases analyzed in the SGS analysis.
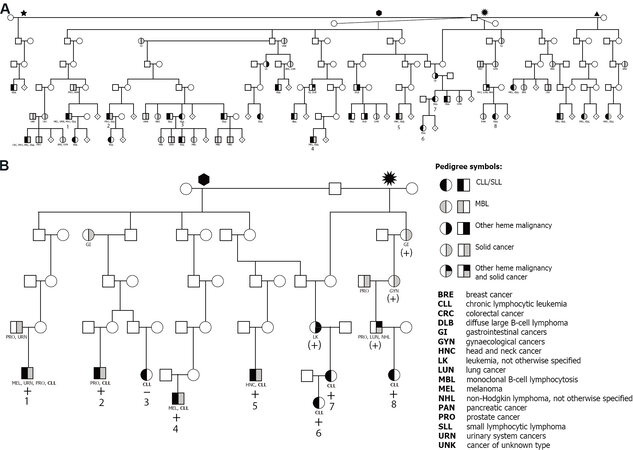
Figure 1. Extended high-risk chronic lymphocytic leukemia (CLL) pedigree from the Utah Population Database. (A) Full extent of CLL cases captured from the common ancestor, with four wives. (B) Reduced pedigree, terminating at each sampled CLL case and only containing the intermediate connecting relatives. “+” indicates sharing of the significant shared 2q22.1 region; “(+)” indicates obligate sharing with a heme malignancy or solid cancer; and “-” indicates non-sharing.
Acquisition of materials and genotyping
Peripheral blood samples were processed to DNA. Individuals in the pedigrees were genotyped using the Illumina Human 610Q high-density single nucleotide polymorphism (SNP) array. Genotypes were called using standard Illumina protocols. Alleles were re-oriented to align with 1000 Genomes Project sequence data[18]. SNP quality control was performed alongside other project data using PLINK and included: SNP call-rate (95%), sample call-rate (90%), removal of monomorphic SNPs, and failure of Hardy-Weinberg equilibrium (P < 1.0 × 10-5)[19-22]. After quality control, 555,091 autosomal SNPs were available for SGS. The average age at diagnosis for these eight sampled cases was 61.5 (min 46, max 72). The average overall survival time for the five cases who subsequently died was 11.2 years (min 4.8, max 15.9).
Shared genomic segment analysis
A detailed explanation of the SGS method, including optimization over subsets and determination of the genome-wide significance threshold has been described elsewhere[15]. Briefly, a segment is defined as the stretch of consecutive SNPs shared IBS by a subset of cases. Sharing is assessed for each subset (n ≥ 2) of cases in the pedigree. A segment is broken when two cases in a subset are opposite homozygotes and thus cannot share. At each position across the genome, the optimal segment across subsets is determined (smallest P-value). Together these become the genome-wide optimal results.
Nominal significance for each segment is established empirically. Simulated genotype configurations under the null hypothesis of no linkage are generated using a gene-drop procedure. This involves random assignment of chromosomes to pedigree founders (individuals in the pedigree without parents) based on a linkage disequilibrium map from the 1000G using graphical modeling[23]. The principles of Mendelian inheritance are then used to “drop” the chromosomes through the pedigree structure with recombination occurring according to the Rutgers genetic map[24]. For each set of simulated genotypes, the SGS sharing is determined and optimal null segments across the genome established in parallel process to that performed in the real data. The nominal empirical P-value for an observed segment is the proportion of null optimal segments at the same position that are the same or longer than that observed. At one million simulations, a distribution is fit, based on the set of genome-wide empirical P-values (under the assumption that the majority of segments across the genome represent the null). This distribution is used to establish the pedigree-specific genome-wide threshold, corresponding to a false-positive rate of μ = 0.05 per genome, based on the Theory of Large Deviations[25]. Simulations then continue, as necessary, until all P-values are estimated to resolution.
Establishing germline sharing
The DNA studied was derived from whole blood lymphocytes and therefore may be contaminated with malignant CLL cells. To delineate possible contamination, we obtained second blood draws for two of the CLL cases and used flow cytometry to cell-sort CD19+/CD5+ cells (malignant CLL cells) and non-malignant cells (reflective of germline). Genotypes from these sorted cells were used to confirm that alleles shared in SGS regions were germline in origin.
Haplotype estimation
At a locus, SGS analysis identifies the region with the best statistical evidence (lowest P-value) and defines the subset of cases that share it (the sharing group). By definition, all cases in the sharing group can share a haplotype across the best region. Subsets of the sharing group may, however, share longer regions (with less significant P-values). We followed the pattern of P-values as they iteratively diminished to identify the longer segments shared by fewer cases in the sharing group. Cases who are removed from subsequent longer regions indicate loss of the ancestral haplotype, i.e., a recombinant event. In this way, the haplotypes for each individual of the sharing group can be estimated surrounding the SGS region.
Human Protein Atlas transcriptome analysis
We used three publicly available datasets from the Human Protein Atlas (HPA) version 20.0
RESULTS
All eight sampled CLL individuals in the pedigree passed genotyping quality control. The final pedigree for analysis included the eight CLL cases separated by 28 meioses. A genome-wide significance threshold of α = 3.94 × 10-7 was established for the pedigree.
One genome-wide significant SGS region was identified at chromosome 2q22.1 (P = 1.9 × 10-7, LOD-equivalent 5.6) [Figure 2A and B]. This 2q22.1 locus is inherited through 26 meioses to seven of the eight studied CLL cases [Figure 1B]. Two additional obligate carriers (parents) with hematological malignancies also shared the segregating region: non-Hodgkin lymphoma, NOS and leukemia, NOS [Figure 1B]. The region shared by all seven CLL cases contains 204 consecutive SNPs and is 0.9 Mb in length, from 136.1-137.0 Mb (GRCh38). Alleles in the sorted cells confirmed the shared region was germline. Figure 3A illustrates the SGS region and each of the seven estimated haplotypes in the case-sharers at this locus. The shared region encompasses the entire CXCR4 gene, part of the gene, THSD7B (thrombospondin type 1 domain containing 7B), and two unstudied non-coding genes (AC112255.1 and RN7SKP141). The mRNA expression of CXCR4 and THSD7B in 14 relevant tissues, cells and cell-lines from the HPA is shown below each gene [Figure 3B][28]. Expression of CXCR4 was evident in all 14 relevant tissues/cell-lines/cells, and highest in bone marrow and lymph node. Expression of THSD7B was virtually nonexistent in all tissues/cells [Figure 3B].

Figure 2. Shared genomic segment (SGS) analysis results. (A) Manhattan plot of the genome-wide SGS optimal segment P-values. Significant threshold (μ = 0.05) is 3.98 × 10-7. Suggestive threshold (μ = 1.0) is 2.98 × 10-6. (B) SGS segment plot focused on the 50 Mb surrounding the significant peak at 2q22.1. The plot shows all the SGS segments and their P-values. Segments in the optimal set (segments that are the most significant P-value at a position in the genome) are highlighted in red.
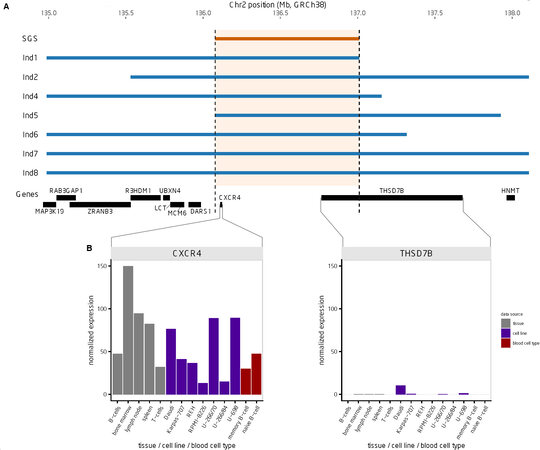
Figure 3. Characterization of the shared genomic segment (SGS) region (A) SGS region, seven estimated haplotypes (inherited from the common founder), and location of genes in the region. (B) Expression of CXCR4 and THSD7B using data from the Human Protein Atlas for 14 relevant tissues/cell lines/blood cell types.
DISCUSSION
Despite the consistent and significant evidence for familial clustering in CLL, the rarity of the malignancy and its etiologic complexity have challenged discovery of segregating risk genes in families. Early linkage studies did not identify any significant loci[5,6,8,9]. The largest collaborative study including 206 mostly nuclear families identified one significant locus (P = 7.7 × 10-5)[7]. With many small pedigrees, a 1-LOD support interval surrounding the peak is standard for localization, identifying the region that has odds within an order of magnitude of the peak. This identifies chromosome 2.2q21.2-2q22.1 as the localized region. This ~6.25 Mb region harbors 18 protein-coding genes (GRCh38) including, as noted by the authors, CXCR4 as the likely candidate. Our study of a large high-risk CLL pedigree represents the largest single pedigree studied to-date. We identified one genome-wide significant finding (P = 1.9 × 10-7), a 0.9 Mb region that lies within the previously suggested ~6.25 Mb region. Our result replicates and substantially narrows the locus, which now implicates only two genes: CXCR4 and THSD7B.
Overlay of expression in relevant tissues point to CXCR4 as the compelling candidate [Figure 3B]. Further, there is a rapidly growing literature on CXCR4 in CLL, while in contrast no published articles exist for THSD7B and CLL. CXCR4 has been shown to be overexpressed in malignant CLL cells[30], and has been associated with disease progression[31,32], and Rai stage[33] as well as worse prognosis[34] and survival in familial CLL[35]. The 5’ UTR of CXCR4 has been shown to be recurrently mutated in CLL[36] and has been found as a proto-onco fusion gene with MAML2[37]. It been additionally been shown to be a key molecule in CLL cell trafficking into and out of the bone marrow[38], referred to as “CXCR4-mediated migration”, and influential in dependencies with the microenvironment[39]. Given its vital function in CLL proliferation, targeting CXCR4 in CLL has shown efficacy in treating the disease as well as modifying drug-response, particularly with the drug ibrutinib[40-45]. Additionally, responses to ibrutinib were influenced by somatic MYD88 and CXCR4 mutations in patients with Waldenström’s macroglobulinemia[46].
In addition to CLL, CXCR4 is overexpressed in over 23 cancers, including lung, prostate, melanoma, and uterine cancers (reviewed in[47,48]). Four of the CLL SGS sharers were also diagnosed with solid cancers: two melanoma, two prostate, urinary systems, and head and neck cancer [Figure 1B]. Three obligate carriers were also diagnosed with solid cancers: prostate, gastrointestinal, gynaecological, and lung cancer [Figure 1B]. The only published article with THSD7B and any cancer is a GWAS study that identified a common variant (rs13405020, P < 7 × 10-6) outside of the SGS region in THSD7B in Korean patients with non-small cell lung cancer[49].
A small case-control gene-panel sequencing study of sporadic CLL included CXCR4, and identified a common variant in CXCR4 (rs2228014, MAF = 0.04) that was increased in CLL cases (uncorrected P = 0.0015)[50]. The association of this variant with CLL was not replicated in a second, larger case-control study (P = 0.84)[51]. However, one rare truncating variant and one missense variant were observed in CXCR4 in two CLL cases with positive family history which was absent from controls (not statistically significant)[51]. Truncating germline mutations in the C-terminus of CXCR4 have been shown to act as gain-of-function mutations and cause WHIM syndrome (warts, hypogammaglobulinemia, infections, and myelokathexis), and Waldenström’s macroglobulinemia[52].
Our analysis was limited to the autosome, a restriction of the current SGS algorithm. None of the six previously proposed CLL risk genes from direct-to-sequencing or CNV analyses in family-based designs are located on the sex chromosomes. These are ITGB2[10], POT1, TERF2IP, ACD[11], DLEU7 and IRF4[13]. All remain to be replicated. Attempts to replicate recurrence of ITGB2 rs2230531 in families[12] or association in sporadic case-control designs have not been successful[53]. We did not find significant or suggestive evidence of segregation of any of these loci in our pedigree, although we are limited by investigating only one family.
In summary, we have studied a single six-generation high-risk CLL pedigree and identified a genome-wide significant region at 2q22.1 shared by seven CLL cases and two obligate carriers with hematological malignancies. The 0.9 Mb region replicates and narrows a previously proposed linkage locus for CLL. In a complex field which has lacked in replication of family-based findings thus far, this result is extremely encouraging. Within the shared region, CXCR4 is a compelling candidate. The seven haplotype carriers in the pedigree provide a valuable resource for pursuing the functionally relevant variant/s (coding or regulatory) that reside on the shared haplotype. Future work will elucidate if, in fact, CXCR4 plays a role in inherited risk, as implicated here.
DECLARATIONS
AcknowledgmentsWe thank the participants and their families who make this research possible. Data collection was made possible, in part, by the Utah Population Database (UPDB) and the Utah Cancer Registry (UCR). Computations were supported by the University of Utah’s Center for High Performance Computing. We thank the University of Utah Health Science Center Flow Cytometry and Genomics Cores, and the staff at the UPDB for their support in the identification of the CLL pedigrees. We greatly appreciate Rob Sargent for technical and programming support performing the SGS analyses.
Authors’ contributionsDesigned the study and wrote the manuscript: Feusier JE, Camp NJ
Contributed to method development: Feusier JE, Madsen MJ, Avery BJ, Camp NJ
Performed data curation and processing: Madsen MJ, Williams JA
Performed analyses: Feusier JE, Madsen MJ, Avery BJ
Provided biological and clinical perspective: Stephens DM, Hu B, Osman AEG, Glenn MJ,
Figures and tables were generated: Feusier JE, Madsen MJ, Avery BJ
Reviewed and edited the manuscript: Feusier JE, Madsen MJ, Avery BJ, Williams JA, Stephens DM, Hu B, Osman AEG, Glenn MJ, Camp NJ
Availability of data and materialsThe Shared Genome Segment (SGS) analysis software is freely available and can be accessed online: https://uofuhealth.utah.edu/huntsman/labs/camp/analysis-tool/shared-genomic-segment.php. Data used in the SGS analysis includes pedigree structures, CLL diagnoses, and genome-wide SNP genotypes. Pedigree structures necessary for these analyses were acquired from the UPDB. These are considered potentially identifiable by the Resource for Genetic and Epidemiologic Research (RGE) - the ethical oversight committee for the UPDB. As a result, access to these data requires review by the RGE committee (contact Jahn Barlow,
The study was made possible by National Cancer Institute (NCI) R01 CA134674 (to NJC). JF was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1 TR002538 and TL1 TR002540. This research was supported by the UCR, which is funded by the NCI's SEER Program, Contract No. HHSN261201800016I, the US Center for Disease Control and Prevention's National Program of Cancer Registries, Cooperative Agreement No. NU58DP0063200, with additional support from the University of Utah and Huntsman Cancer Foundation. Partial support for all datasets within the UPDB is provided by the University of Utah, Huntsman Cancer Institute and the Huntsman Cancer Institute Cancer Center Support grant, P30 CA42014 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of interestStephens DM has served on advisory boards for Beigene, Jannsen, Pharmacyclics, Epizyme, Adaptive, TG Therapeutics, Karyopharm, Innate. Stephens DM has received research funding from Karyopharm, Acerta, Arqule, Mingsight, Juno, Gilead, Verastem. Hu B has received research funding from Miragen, Roche, CRISPR Therapeutics and Celgene.
Ethical approval and consent to participateAll work was performed under Approved University of Utah IRB protocol 88405. Ethics committees at the University of Utah approved this research. All participants provided written informed consent.
Consent for publicationNot Applicable.
Copyright© The Author(s) 2021
REFERENCES
1. Chronic lymphocytic leukemia - Cancer Stat Facts. Available from: https://seer.cancer.gov/statfacts/html/clyl.html. [Last accessed on 21 May 2021].
2. Goldin LR, Pfeiffer RM, Li X, Hemminki K. Familial risk of lymphoproliferative tumors in families of patients with chronic lymphocytic leukemia: results from the Swedish Family-Cancer Database. Blood 2004;104:1850-4.
3. Kerber RA, O'Brien E. A cohort study of cancer risk in relation to family histories of cancer in the Utah population database. Cancer 2005;103:1906-15.
4. Slager SL, Caporaso NE, de Sanjose S, Goldin LR. Genetic susceptibility to chronic lymphocytic leukemia. Semin Hematol 2013;50:296-302.
5. Goldin LR, Ishibe N, Sgambati M, et al. A genome scan of 18 families with chronic lymphocytic leukaemia. Br J Haematol 2003;121:866-73.
6. Sellick GS, Webb EL, Allinson R, et al. A high-density SNP genomewide linkage scan for chronic lymphocytic leukemia-susceptibility loci. Am J Hum Genet 2005;77:420-9.
7. Sellick GS, Goldin LR, Wild RW, et al. A high-density SNP genome-wide linkage search of 206 families identifies susceptibility loci for chronic lymphocytic leukemia. Blood 2007;110:3326-33.
8. Raval A, Tanner SM, Byrd JC, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007;129:879-90.
9. Fuller SJ, Papaemmanuil E, McKinnon L, et al. Analysis of a large multi-generational family provides insight into the genetics of chronic lymphocytic leukemia. Br J Haematol 2008;142:238-45.
10. Goldin LR, McMaster ML, Rotunno M, et al. Whole exome sequencing in families with CLL detects a variant in Integrin β 2 associated with disease susceptibility. Blood 2016;128:2261-3.
11. Speedy HE, Kinnersley B, Chubb D, et al. Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood 2016;128:2319-26.
12. Blackburn NB, Marthick JR, Banks A, et al. Evaluating a CLL susceptibility variant in ITGB2 in families with multiple subtypes of hematological malignancies. Blood 2017;130:86-8.
13. Brown JR, Hanna M, Tesar B, et al. Germline copy number variation associated with Mendelian inheritance of CLL in two families. Leukemia 2012;26:1710-3.
14. Hanson HA, Leiser CL, Madsen MJ, et al. Family study designs informed by tumor heterogeneity and multi-cancer pleiotropies: the power of the Utah Population Database. Cancer Epidemiol Biomarkers Prev 2020;29:807-15.
15. Waller RG, Darlington TM, Wei X, et al. Novel pedigree analysis implicates DNA repair and chromatin remodeling in multiple myeloma risk. PLoS Genet 2018;14:e1007111.
16. Glenn MJ, Madsen MJ, Davis E, et al. Elevated IgM and abnormal free light chain ratio are increased in relatives from high-risk chronic lymphocytic leukemia pedigrees. Blood Cancer J 2019;9:25.
17. Knight S, Abo RP, Abel HJ, et al. Shared genomic segment analysis: the power to find rare disease variants. Ann Hum Genet 2012;76:500-9.
18. Auton A, Brooks LD, Durbin RM. The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015;526:68-74.
19. Purcell S, Chang C. PLINK 1.9. Available from: https://www.cog-genomics.org/plink/1.9/. [Last accessed on 21 May 2021].
20. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7.
21. Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 2005;76:887-93.
22. Graffelman J, Moreno V. The mid p-value in exact tests for Hardy-Weinberg equilibrium. Stat Appl Genet Mol Biol 2013;12:433-48.
23. Thomas A, Camp NJ, Farnham JM, Allen-Brady K, Cannon-Albright LA. Shared genomic segment analysis. Mapping disease predisposition genes in extended pedigrees using SNP genotype assays. Ann Hum Genet 2008;72:279-87.
24. Matise TC, Chen F, Chen W, et al. A second-generation combined linkage physical map of the human genome. Genome Res 2007;17:1783-6.
25. Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 1995;11:241-7.
26. Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419.
27. Thul PJ, Åkesson L, Wiking M, et al. A subcellular map of the human proteome. Science 2017;356:eaal3321.
28. The Human Protein Atlas. Available from: https://www.proteinatlas.org/. [Last accessed 21 May 2021].
29. Assays and annotation - The Human Protein Atlas. Available from: https://www.proteinatlas.org/about/assays+annotation#hpa_rna. [Last accessed 21 May 2021].
30. Möhle R, Failenschmid C, Bautz F, Kanz L. Overexpression of the chemokine receptor CXCR4 in B cell chronic lymphocytic leukemia is associated with increased functional response to stromal cell-derived factor-1 (SDF-1). Leukemia 1999;13:1954-9.
31. Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma 2002;43:461-6.
32. Schmidt J, Federmann B, Schindler N, et al. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol 2015;169:795-803.
33. Ghobrial IM, Bone ND, Stenson MJ, et al. Expression of the chemokine receptors CXCR4 and CCR7 and disease progression in B-cell chronic lymphocytic leukemia/ small lymphocytic lymphoma. Mayo Clin Proc 2004;79:318-25.
34. Ganghammer S, Gutjahr J, Hutterer E, et al. Combined CXCR3/CXCR4 measurements are of high prognostic value in chronic lymphocytic leukemia due to negative co-operativity of the receptors. Haematologica 2016;101:e99-102.
35. Ishibe N, Albitar M, Jilani IB, Goldin LR, Marti GE, Caporaso NE. CXCR4 expression is associated with survival in familial chronic lymphocytic leukemia, but CD38 expression is not. Blood 2002;100:1100-1.
36. Puente XS, Beà S, Valdés-Mas R, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015;526:519-24.
37. Acunzo M, Romano G, Wernicke D, et al. Translocation t(2;11) in CLL cells results in CXCR4/MAML2 fusion oncogene. Blood 2014;124:259-62.
38. Redondo-Muñoz J, García-Pardo A, Teixidó J. Molecular players in hematologic tumor cell trafficking. Front Immunol 2019;10:156.
39. Kriston C, Plander M, Márk Á, et al. In contrast to high CD49d, low CXCR4 expression indicates the dependency of chronic lymphocytic leukemia (CLL) cells on the microenvironment. Ann Hematol 2018;97:2145-52.
40. Hacken E, Burger JA. Microenvironment dependency in Chronic Lymphocytic Leukemia: The basis for new targeted therapies. Pharmacol Ther 2014;144:338-48.
41. Pavlasova G, Borsky M, Seda V, et al. Ibrutinib inhibits CD20 upregulation on CLL B cells mediated by the CXCR4/SDF-1 axis. Blood 2016;128:1609-13.
42. Martini V, Gattazzo C, Frezzato F, et al. Cortactin, a Lyn substrate, is a checkpoint molecule at the intersection of BCR and CXCR4 signalling pathway in chronic lymphocytic leukaemia cells. Br J Haematol 2017;178:81-93.
43. Kashyap MK, Kumar D, Jones H, et al. Ulocuplumab (BMS-936564 / MDX1338): a fully human anti-CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen species-dependent pathway. Oncotarget 2016;7:2809-22.
44. Secchiero P, Voltan R, Rimondi E, et al. The γ-secretase inhibitors enhance the anti-leukemic activity of ibrutinib in B-CLL cells. Oncotarget 2017;8:59235-45.
45. Shaim H, Estrov Z, Harris D, et al. The CXCR4-STAT3-IL-10 pathway controls the immunoregulatory function of chronic lymphocytic leukemia and is modulated by lenalidomide. Front Immunol 2017;8:1773.
46. Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenström's macroglobulinemia. N Engl J Med 2015;372:1430-40.
47. Chatterjee S, Behnam Azad B, Nimmagadda S. . The intricate role of CXCR4 in cancer. In: Pomper MG, Fisher PB, editors. Emerging applications of molecular imaging to oncology. Amsterdam: Elsevier; 2014. p. 31-82.
48. Scala S. Molecular pathways: targeting the CXCR4-CXCL12 axis--untapped potential in the tumor microenvironment. Clin Cancer Res 2015;21:4278-85.
49. Lee Y, Yoon KA, Joo J, et al. Prognostic implications of genetic variants in advanced non-small cell lung cancer: a genome-wide association study. Carcinogenesis 2013;34:307-13.
50. Enjuanes A, Benavente Y, Bosch F, et al. Genetic variants in apoptosis and immunoregulation-related genes are associated with risk of chronic lymphocytic leukemia. Cancer Res 2008;68:10178-86.
51. Crowther-Swanepoel D, Qureshi M, Dyer MJ, et al. Genetic variation in CXCR4 and risk of chronic lymphocytic leukemia. Blood 2009;114:4843-6.
52. Milanesi S, Locati M, Borroni EM. Aberrant CXCR4 signaling at crossroad of WHIM syndrome and Waldenstrom's macroglobulinemia. Int J Mol Sci 2020;21:5696.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Feusier JE, Madsen MJ, Avery BJ, Williams JA, Stephens DM, Hu B, Osman AEG, Glenn MJ, Camp NJ. Shared genomic segment analysis in a large high-risk chronic lymphocytic leukemia pedigree implicates CXCR4 in inherited risk. J Transl Genet Genom 2021;5:189-99. http://dx.doi.org/10.20517/jtgg.2021.05
AMA Style
Feusier JE, Madsen MJ, Avery BJ, Williams JA, Stephens DM, Hu B, Osman AEG, Glenn MJ, Camp NJ. Shared genomic segment analysis in a large high-risk chronic lymphocytic leukemia pedigree implicates CXCR4 in inherited risk. Journal of Translational Genetics and Genomics. 2021; 5(2): 189-99. http://dx.doi.org/10.20517/jtgg.2021.05
Chicago/Turabian Style
Feusier, Julie E., Michael J. Madsen, Brian J. Avery, Justin A. Williams, Deborah M. Stephens, Boyu Hu, Afaf E. G. Osman, Martha J. Glenn, Nicola J. Camp. 2021. "Shared genomic segment analysis in a large high-risk chronic lymphocytic leukemia pedigree implicates CXCR4 in inherited risk" Journal of Translational Genetics and Genomics. 5, no.2: 189-99. http://dx.doi.org/10.20517/jtgg.2021.05
ACS Style
Feusier, JE.; Madsen MJ.; Avery BJ.; Williams JA.; Stephens DM.; Hu B.; Osman AEG.; Glenn MJ.; Camp NJ. Shared genomic segment analysis in a large high-risk chronic lymphocytic leukemia pedigree implicates CXCR4 in inherited risk. J. Transl. Genet. Genom. 2021, 5, 189-99. http://dx.doi.org/10.20517/jtgg.2021.05
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 12
likes
Like This Article 12
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.