
Genetics and genomics of extranodal natural killer/T cell lymphoma: from etiology to treatment
 0
0Abstract
Extranodal natural killer/T cell lymphoma (NKTCL) is a heterogenous and unique epidemiological non-Hodgkin’s lymphoma, which is strongly associated with Epstein-Barr virus (EBV) infection. Based on the development of various sequencing methods and molecular biology technologies, genome- and transcriptome-wide association studies of NKTCL have provided insight into the etiology and pathogenesis of NKTCL. Comparative genomic hybridization detected variations in tumor suppressor genes such as PRDM1, RUNX3, and EZH2. Whole-exome sequencing identified pathogenic variant such as DDX3X, and TP53. Signal pathways such as the Janus kinase/signal transduction and activator of transcription pathway and nuclear factor kappaB pathway are frequently abnormal in NKTCL. In addition, programmed death-1, programmed death ligand-1, and the human leukocyte antigen risk alleles are significantly associated with NKTCL pathogenesis. Meanwhile, epigenetics analysis has also exposited changes such as PTPRK, HACE1, microRNAs, and long non-coding RNAs, which play important role on the development and biology of NKTCL. EBV infection is tightly correlated with NKTCL. Viral genomic alterations and lytic genes of EBV are reported to have pathogenic effects on host cells that contribute to the etiology of NKTCL. We summarize the genomic and genetic alterations during the pathogenesis and development of NKTCL and exhibit the potential therapeutic targets that are worth exploring in future research and clinical trials.
Keywords
INTRODUCTION
Extranodal natural killer/T cell lymphoma (NKTCL) commonly involves the upper aerodigestive tract such as the nasal cavity and is prevalent in Asia, Mexico, and Latin America[1,2] while rare in the United States and Europe[2,3]. NKTCL was previously called “lethal midline granuloma”, of which the pathological features are pleomorphic and atypical cells ranging from medium to large, obvious necrosis, irregular nucleus and cytoplasmic changes, and showing an angiocentric/angiodestructive growth pattern with infiltration of immune inflammatory cells[1,4-7]. Neoplasm cells are positive for CD56, CD2, and CD3ε with the expression of cytotoxic molecules, such as TIA1 and Granzyme B. Epstein-Barr virus (EBV) DNA and non-coding EBV-encoded RNAs could be detected in tumor tissues[8-10], which leads to the correlation of EBV infection and the pathogenesis of NKTCL. In addition, NKTCL tumor cells express P-glycoprotein, which is related to multidrug resistance, resulting in poor response to anthracyclines[11]. Concurrent L-asparaginase-containing regimens with or without radiotherapy are the current standard treatment. The International T-cell Project’s report showed great improvement in the survival of NKTCL patients over the past decade[12]. The five-year overall survival rates for stage I, stage II, and stage III-IV are 55%, 42%, and 24%, respectively[12]. However, the treatment for advanced-stage, relapsed, or refractory patients is still challenging[12]. Therefore, there is an urgent need for more effective treatments to improve the survival of NKTCL patients.
Identifying the unique gene expression profile, dysregulated molecules, and signaling pathways of NKTCL provided a new perception for understanding pathogenic mechanisms and potential therapeutic targets[13,14]. Genome-wide association study revealed susceptible genes significantly associated with the higher risk of NKTCL[15,16]. EBV genome and transcriptomics analysis showed that EBV infection might contribute to the pathogenesis of NKTCL by affecting the host cell genome[17]. Exploring the genomics and genetic variations in the tumorigenesis and development of NKTCL from the molecular aspects has vital biological significance for the future clinical diagnosis, treatment, and prognosis of NKTCL. Xiong et al.[18] integrated the cell origin, EBV expression pattern, clinical significance and characteristics of alterations in the human genome of NKTCL samples, and divided NKTCL into three molecular subtypes: TSIM subtype [based on variants in the Janus kinase/signal transduction and activator of transcription (JAK/STAT) pathway and TP53], MB subtype (based on MGA mutation and 1p22.1/BRDT loss of heterozygosity), and HEA subtype (based on HDAC9, EP300, and ARID1A mutation)[18]. Disease stratification bridges the pathogenesis of NKTCL with clinical intervention from pathogenesis theory into clinical practice.
THE ALTERATION OF HUMAN GENETICS AND POTENTIAL TREATMENTS
The network of NKTCL pathogenesis has not been totally revealed. However, changes in a variety of genes and signaling pathways have been reported. We try to excavate underlying signatures from reported genetic variations and search the genetic hallmarks increasing the risk of NKTCL, which furnish guidance for mechanism-based anti-tumor therapy.
Tumor suppressor genes and Somatic mutations
Dysfunction of tumor suppressor genes occupies a significant position in the development of hematological malignancies, which has been discussed in leukemia and myeloma[19,20]. Many major candidate genes, such as PRDM1, TP53, HACE1, FOXO3, and ATG5, are located in the deletion region of chromosome 6q21, which is the most frequently discussed in NKTCL[13,14,21]. There are recurrent somatic mutations in NKTCL as well.
PRDM1
PRDM1 encodes Blimp, a transcription inhibitor of NK cell and T cell differentiation, and regulates the proliferation and maturation of NK cells[22]. As a tumor suppressor gene (TSG) located in the 6q21 region, PRDM1 is inactivated due to mutation, promoter methylation, or deletion, with a low expression level in NKTCL[23,24]. Inhibition or inactivation of PRDM1 is supposed to upregulate genes or pathways related to proliferation and cell cycle regulation such as MYC and to downregulate the pro-apoptotic factor BIM[21]. The above indicates that PRDM1 may mediate malignant transformation of cells and be a potential genetic target. A recent study found that pan-acetyltransferase inhibitor vorinostat might restore PRDM1 response to IL21 through decreasing BCL6 bound to PRDM1 in follicular lymphoma (FL) cells[25]. According to the results from vorinostat on nonfunctional CREBBP, FL cells showed a significant increase in PRDM1 expression after IL21 exposure. The expression of PRDM1 might be an important response predictor for pan-HDAC inhibitors on FL cells. However, whether agents that restore PRDM1 exert anti-tumor activity in NKTCL is still to be explored.
RUNX3 and MYC
Runt-related transcription factor 3 (RUNX3) presents a bilateral significance in different tumor backgrounds. It is overexpressed in colorectal cancer and could promote TRAIL-induced apoptosis exerting anti-tumor effect[26]. In cutaneous T-cell lymphoma, the re-expression of RUNX3 decreases tumor cell survival and induces apoptosis, indicating that RUNX3 acts as tumor suppressor gene[27].
There are different degrees of expression of MYC in different types of lymphoma[28]. Compared with NK cells, MYC is highly expressed and activated in NKTCL. The inhibition of its target genes may be a possible mechanism for it playing an important role in the development of NKTCL[29].
Selvarajan et al.[30] demonstrated that RUNX3 is overexpressed and oncogenic in NKTCL. MYC is involved in the positive transcriptional regulation of RUNX3[30]. JQ1, a small molecule inhibitor, may induce apoptosis of NKTCL cell lines by inhibiting transcription of MYC and downregulating RUNX3, which indicates that RUNX3 and MYC may be potential therapeutic targets in NKTCL[30]. In addition, homoharringtonine is considered to downregulate the expression of MYC via directly binding to NKRF, the inhibitor of nuclear factor kappaB (NF-κB)[31]. Relevant clinical trials have shown that homoharringtonine-based induction regimens can significantly improve the complete remission (CR) rate and progression-free survival of acute myeloid leukemia patients[32]. The potential therapeutic implication of this reagent deserves to be further explored in NKTCL.
EZH2
Enhancer of zeste homolog 2 (EZH2) is overexpressed in NKTCL and acts as oncogene in tumor progression[33,34]. Previous studies have shown that EZH2 undergoes frequently somatic mutations in FL and diffuse large B-cell lymphoma (DLBCL), resulting in dysregulation of epigenetic silence function of methyltransferase[35]. Interestingly, Yan et al.[36] considered that EZH2, as a transcriptional activator, promoted proliferation through regulation of cyclin D1 expression by a non-canonical pathway. JAK3 kinase inhibitor PF956980 can induce the arrest of EZH2 phosphorylation and block its non-canonical pathway via limiting the growth advantage of NKTCL cells[36]. A recent study found that MELK (maternal embryonic leucine zipper kinase), which regulates the ubiquitination and turnover of EZH2, increased EZH2 S220 phosphorylation and promoted stabilization of EZH2 protein in NKTCL[37]. These studies demonstrate a potential target role of EZH2, and a JAK3 inhibitor may be a prospective treatment option. Additionally, a phase II clinical trial (Clinical Trial No. NCT01897571) about the EZH2 inhibitor tazemetostat treating refractory/relapsed B-cell non-Hodgkin’s lymphoma patients is ongoing. Of note, results from a phase I clinical trial demonstrated that tazemetostat had a favorable safety profile and anti-tumor activity in refractory NHL and advanced solid tumors, including epithelioid sarcoma[38]. Analogously, EZH2 inhibitors might be worth exploring in NKTCL treatment.
DDX3X
DDX3X belongs to the RNA helicase family, and its gene mutation is involved in the formation of a variety of human tumors[39-41]. According to Jiang et al.[42], DDX3X is the most common somatic mutation gene (20%, 21/105) in NKTCL. Notably, tumors with DDX3X mutation show activation and upregulation of NF-κB and mitogen-activated protein kinase (MAPK) pathways, which also reflects that DDX3X mutation has biological significance in NKTCL pathogenic process. However, the actual significance in clinical treatment of NKTCL still needs more studies and trials to confirm.
Other TSGs and Somatic mutations
The tumor suppressor gene TP53 is considered to be dysfunctional due to somatic mutations[41,43]. Genes normally suppressed by TP53 are upregulated in tumor tissues, which may lead to the progression of NKTCL[29]. FOXO3 is lowly expressed in NKTCL and induces apoptosis and cell cycle arrest of NK cell lines, which is recognized to be of great significance in the pathogenesis of NKTCL[23]. Hexokinase domain component 1 has been proven to be transcriptionally upregulated in the NKTCL cell line. It not only promotes the proliferation of tumor cells but also inhibits EBV replication and P-glycoprotein expression through promoting the overproduction of ROS and DNA damage[44]. Besides, survivin is overexpressed in NKTCL. In vitro, terameprocol, a survivin inhibitor, can significantly inhibit the growth of NKTCL cell lines[29].
The alterations of these genes further enrich the molecular network of NKTCL pathogenesis and provide potent targets for anti-tumor therapy. The relevant content is summarized in Table 1. Whether they could be used as valuable clinical factors for diagnosis and treatment needs to be verified in larger-scale experiments.
Key targets and their potential pathogenic mechanisms and therapeutic significance from the alterations TSGs and somatic mutations in NKTCL
| The alterations of human genetics | Potential hallmark/signaling pathways | Role in pathogenic mechanism of lymphoma | Ref. | Potential treatment significance | Ref. |
| TSGs and somatic mutations | PRDM1 | Downregulated and promotes cell proliferation and reduces apoptosis | Karube et al.[23] Küçük et al.[24] | IL21 plus vorinostat upregulates PRDM1 expression in FL and may be explored in NKTCL treatment | Desmots et al.[25] |
| RUNX3 | Overexpressed and oncogenic in NKTCL | Selvarajan et al.[30] | JQ1, a molecule inhibitor, may induce apoptosis in vitro | Selvarajan et al.[30] | |
| EZH2 | Overexpressed and promotes cell proliferation as a transcriptional activator by a non-canonical pathway | Yan et al.[36] | JAK3 inhibitor PF956980 blocks the non-canonical pathway of EZH2 and limit cell growth in vitro A phase II clinical trial about EZH2 inhibitor tazemetostat treating NHL is ongoing, and it may try to be included in NKTCL therapy | Yan et al.[36] Italiano et al.[38] | |
| MYC | Highly expressed and inhibits its target genes | Ng et al.[29] | HHT improved the CR and PFS of AML cases in a clinical trial and deserves to be further explored in NKTCL | Jin et al.[32] | |
| DDX3X | DDX3X mutation has vital significance in lymphoma pathogenesis | Jiang et al.[42] | N/A | N/A | |
| P53 | Dysfunctional and may lead to the progression of NKTCL | Ng et al.[29] Choi et al.[43] | N/A | N/A | |
| FOXO3 | Downregulated and induces apoptosis and cell cycle arrest in vitro. | Karube et al.[23] | N/A | N/A | |
| HKDC1 | Upregulated, promotes the proliferation of tumor cells, and inhibits EBV replication and P-gp expression. | Chen et al.[44] | N/A | N/A | |
| Survivin | Upregulated and inhibits apoptosis in NKTCL. | Ng et al.[29] | Terameprool, as a survivin inhibitor, inhibits the survival of NKTCL cell lines | Ng et al.[29] |
Oncogenic signaling pathways
JAK/STAT signaling pathway
Targeted capture sequencing observed somatic alteration of the JAK/STAT pathway in 78% (85/109) of NKTCL samples. STAT3 and TP53 genes (21%, 23/109) were mutated most frequently, followed by JAK3, JAK1, and suppressor of cytokine signaling 1 mutations. Furthermore, STAT3 activation resulting from mutations or abnormal phosphorylation may drive the high expression of programmed death ligand-1 (PD-L1), which may have an influence on immune escape of NKTCL[45]. In another study, STAT3 is the most common mutant gene of NKTCL (9/34, 26.5%), and all mutations are located in the SRC homology 2 domain that seems to be an underlying effective target region[46]. Apart from mutations, abnormal expression of phosphorylated STAT3 (pSTAT3) at Tyr705 is also recognized as aberrant activation signature[13]. Dysregulated JAK2 mediates the constitutive phosphorylation of STAT3 (Tyr705) facilitating the growth of NKTCL MEC04 cells and Ser727 phosphorylation of activated MAP-Kinase/Erk pathway, both of which have similar oncogenic significance[47]. In addition, receptor-type tyrosine-protein phosphatase κ (PTPRK) binding to pSTAT3 led to the dephosphorylation of pSTAT3, and some NKTCL patients suffer from function deficiency of PTPRK according to the previous formulation[48]. The above suggests that STAT3 is activated through different mechanisms in NKTCL.
STAT3 obtained phosphorylated activation following JAK3 constitutive phosphorylation on tyrosine Tyr980[49]. Whole-exome sequencing detected JAK3 mutations (A572V and A573V) that exceptionally activated the JAK/STAT signaling pathway and had a tumorigenic effect on NKTCL[50]. Moreover,
According to the above studies, the dysregulation of JAK/STAT pathway is highly prevalent in NKTCL and may play an important role in the pathogenesis of the disease through diverse mechanisms. JAK3 and STAT3 are two latent therapeutic targets, and their inhibitors might have promising results in the treatment of NKTCL patients. In the future, the feasibility and safety of the treatment strategy targeting the deregulated JAK/STAT pathway should be further studied.
NF-κB signaling pathway
NF-κB has critical biological implications in the growth, proliferation, differentiation, and regulation of lymphocytes that are regarded as vital pathogenetic factors in lymphomas[53]. Expression of RelA and cRel, two canonical molecules of the NF-κB pathway in NKTCL, suggested abnormal activation of canonical
PDGF signaling pathway
Platelet-derived growth factor alpha (PDGFRα) is overexpressed in NKTCL, and imatinib mesylate, a PDGFR inhibitor, has a limited effect on the growth of the PDGFRα + NKTCL cell line, which indicates that the PDGF pathway was involved in the pathogenic process of NKTCL. However, there was still no evidence explaining the dysregulation of PDGFRα completely[13]. Piccaluga et al.[57] revealed that activated PDGFRα fostered the proliferation of peripheral T-cell lymphomas, not otherwise specified (PTCL or NOS), cells through autocrine loop.
Other oncogenic signaling pathways
Gene sets analysis also discerned other biological pathways in NKTCL including the MAPK, WNT, AKT, and vascular endothelial growth factor (VEGF) signaling pathways[13]. The NOTCH and aurora kinase A (AURKA) pathways were upregulated in NKTCL. Notch inhibitors, which potently inhibited γ-secretase and Notch processing, and an AURKA inhibitor (MK-8745) both may inhibit the proliferation and cell cycle regulation of NK-lymphoma cell lines[58]. The Akt/mammalian/mechanistic target of rapamycin (mTOR) pathway is abnormally activated in EBV-associated T- and NK-cell lymphoma, and their inhibitors restricted effectually proliferation of cell lines[59]. The excessive expression of phosphatidylinositol 3-kinase PIK3 isoforms, containing PIK3α, PIK3β, PIK3γ, and PIK3δ, indicated the abnormally dysregulated activation of PIK3 pathway in NKTCL[60]. VEGF was overexpressed in cutaneous NKTCL and related to poor prognosis. It also provided a potential basis for disorder of the VEGF pathway in NKTCL[61]. The MYC/MAP3K6 pathway is considered to be a characteristic manifestation of MB subtype NKTCL[18].
Integrating the alterations of human genetics in NKTCL, we simply depict an oncogenic molecular network linking dysregulated genes and signaling pathways in Figure 1. We also summarize the relevant content about oncogenic signaling pathways in Table 2.
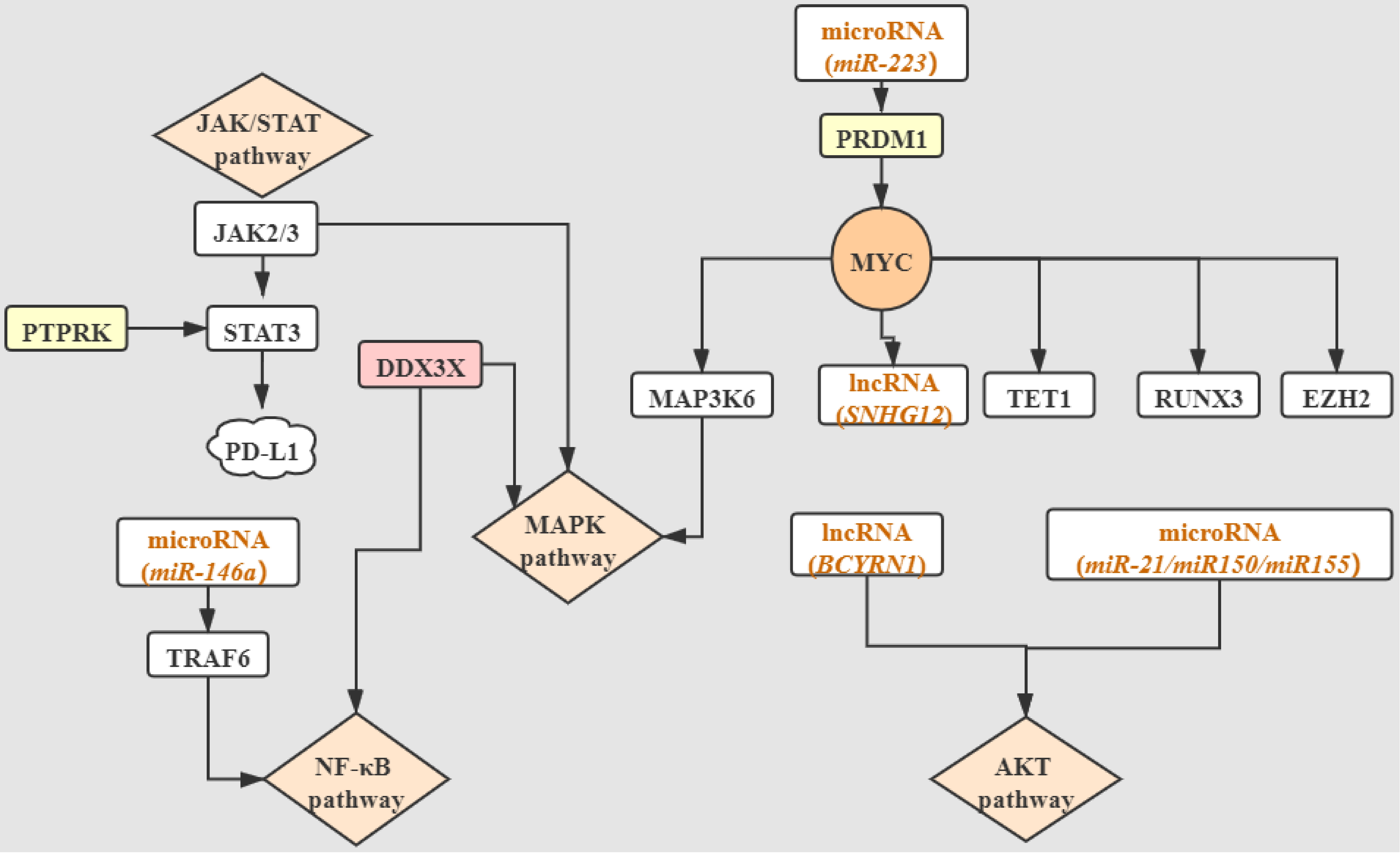
Figure 1. The interrelationships among JAK/STAT, NF-κB, MAPK, and AKT carcinogenic pathways and dysregulated tumor suppressor genes, somatic mutations, and epigenetic mutations. Molecules located at junctions of the non-arrow ends have effects on the part pointed by the arrows. Specific explanations are disclosed in the corresponding sections. JAK/STAT: Janus kinase/signal transduction and activator of transcription; JAK2/3: Janus kinase2/3; STAT3: signal transduction and activator of transcription 3; PTPRK: receptor-type tyrosine-protein phosphatase κ; PDL1: programmed death ligand-1; NK-κB: nuclear factor kappaB; TRAF6: tumor necrosis factor receptor associated factor 6; DDX3X: DEAD-box helicase 3 X-linked; MAPK: mitogen-activated protein kinase; MAP3K6: mitogen-activated protein kinase kinase kinase 6; PRDM1: PR/SET domain 1; MYC: bHLH transcription factor; SNHG12: small nucleolar RNA host gene 12; TET1: methylcytosine dioxygenase ten-eleven translocation 1; RUNX3: runt-related transcription factor 3; EZH2: enhancer of zeste homolog 2; BCYRN1: brain cytoplasmic RNA 1; AKT: protein kinase B.
Vital signaling pathways and their potential oncogenic mechanisms and target significance in NKTCL
| The alterations of human genetics | Potential hallmark/signaling pathways | Role in pathogenic mechanism of lymphoma | Ref. | Potential treatment significance | Ref. |
| Signaling Pathways | JAK/STAT | Aberrant activity via mutations or abnormal phosphorylation | Huang et al.[13] Song et al.[45] Bouchekioua et al.[49] | JAK3 inhibitor: tofacitinib inhibited the growth of cell lines CP-690550 restrained growth and invasion of tumor cells PRN371 apparently inhibited proliferation of NKTCL cells STAT3 inhibitor: stattic inhibited malignant growth advantage in vitro | Bouchekioua et al.[49] Koo et al.[50] Sim et al.[51] Nairismägi et al.[52] |
| NK-κB | Dysregulated and LMP1 induces eIF4E, survivin, and PD-L1 via NK-κB pathway, which may contribute to NKTCL progression | Ng et al.[29] Huang et al.[13] Sun et al.[54] Sun et al.[55] Bi et al.[56] | NF-κB has critical biological implications in NKTCL and targeting the pathway might bring potent clinical value | N/A | |
| PGDF | PDGFRα is overexpressed in NKTCL, and the actual oncogenic sense needs to disclose | Huang et al.[13] | Imatinib mesylate, a PDGFR inhibitor, has a limit effect on cell growth in vitro | Huang et al.[13] | |
| AKT/mTOR | Abnormally activated and promotes cell growth | Kawada et al.[59] | mTOR inhibitors: Rapamycin suppressed mTOR activity and limited cell proliferation in vitro CCI-779 inhibited tumor growth in vivo and in vitro | Kawada et al.[59] | |
| MAPK | MYC/MAP3K6 pathway is a characteristic manifestation of MB subtype NKTCL | Xiong et al.[18] | N/A | N/A | |
| AURKA | Upregulated and may accelerate neoplasm cell proliferation | Iqbal et al.[58] | AURKA inhibitor MK-8745 inhibits proliferation and cell cycle regulation | Iqbal et al.[58] | |
| NOTCH | Upregulated and plays a role in development of neoplasm | Iqbal et al.[58] | NOTCH inhibitors potently inhibit NK-lymphoma cell lines | Iqbal et al.[58] |
Epigenetic variations
PTPRK
PTPRK directly and selectively dephosphorylates the substrate, and loss of phosphatase activity will cause the disruption of cell junctions and enhance invasive characteristics[62]. Because of promoter hypermethylation (16/27, 59%) or monoallelic gene deletion (8/27, 30%), PTPRK expression is frequently downregulated in NKTCL, which is thought to promote constitutive activation of STAT3 and mediate the inhibition of cells proliferation, invasion, and migration. Similarly, demethylation reagent 5-aza-2’-deoxycytidine-induced PTPRK re-expression confirms the aberrant epigenetic changes of PTPRK in NKTCL[48]. PTPRK may be a potential target of NKTCL epigenetic therapy. Whether its promoter methylation and pSTAT3 level can be used as biomarkers for diagnosis or prognosis of NKTCL needs further exploration.
HACE1
HACE1, the novel E3 ubiquitin ligase mapping to the deletion region of 6q21 chromosome, inhibits cell cycle progression via regulating the degradation of cyclin D1. As a tumor suppressor gene, it has been found to be silenced by its CpG methylation in a variety of tumors[63]. Interestingly, another study showed a normal HACE1 protein expression level in the NKTCL cell line but did not observe a similar cell cycle arrest[64]. These inconsistent results indicate that the specific function of HACE1 in NKTCL is not clear, and more studies are needed to discover the real function of HACE1 in NKTCL.
The ten-eleven translocation 1
The expression of the methylcytosine dioxygenase ten-eleven translocation 1 (TET1) maintains DNA hydroxymethylation and prevents the DNA hypermethylation of cells, whose deficiency leads to B-cell lymphoma. Promoter CpG methylation is the main transcriptional silencing mechanism of TET1[65]. However, Poole et al.[66] showed that inactivation of MYC relieved the regulation of TET1 resulting from the overexpression of TET1 in T-cell acute lymphatic leukemia acting as a tumor promoting factor. There was continual methylation of TET1 occurring in 83% (10/12) of NKTCL[67]. The functional mechanism of TET1 in the tumorigenesis of NKTCL has not been completely revealed, but it is indeed a potentially meaningful epigenetic marker.
Other epigenetic variations
The lysine [K]-specific methyltransferase 2D (KMT2D), also known as MLL2, encodes a histone methyltransferase. Mutations of KMT2D may cause dysregulation of gene transcription regulation, which may contribute to the development of NKTCL[43]. Loss of function mutations of BCOR are considered highly specific in NKTCL[41]. Another study demonstrated that BCOR and MLL2 were mutated genes followed only by STAT3[46]. The two aberrant mutated chromatin-modifying genes may also be significant targets of NKTCL epigenetic therapy. All the content regarding epigenetic variations is summarized in Table 3.
Summary of key targets, potential pathogenesis mechanisms, and treatment significance in NKTCL epigenetic variations
| The alterations of human genetics | Potential hallmark/signaling pathways | Role in pathogenic mechanism of lymphoma | Ref. | Potential treatment significance | Ref. |
| Epigenetic variations | PTPRK | Downregulated for methylation or deletion and mediates inhibition of cells proliferation, invasion, and migration | Chen et al.[48] | PTPRK may be a potential target of NKTCL epigenetic therapy | N/A |
| HACE1 | Encodes E3 ubiquitin ligase and inhibits cell cycle progression, but silenced due to CpG methylation The specific function of HACE1 is not clear | Zhang et al.[63] | N/A | N/A | |
| TET1 | Frequently methylated, and the oncogenic mechanism has not been revealed | Li et al.[67] | N/A | N/A | |
| KMT2D | Mutations may cause dysregulation of gene transcription regulation | Choi et al.[43] | N/A | N/A | |
| BCOR | Loss of function mutations of BCOR are considered highly specific in NKTCL | Dobashi et al.[41] | N/A | N/A |
microRNAs and lncRNAs
microRNAs
microRNAs are small non-coding RNAs with a length of 19-24 bp nucleotides that negatively regulate target genes and are associated with the regulation of crucial biological processes such as cell growth, differentiation, and apoptosis[68]. Many studies have revealed the functional significance of microRNAs dysregulation in NKTCL.
Ng et al.[69] discovered that miR-342-5p, miR-26b, miR-363, miR-150, miR28-5p, miR-26a, and miR-101 were downregulated in NKTCL tumor tissues and in vitro, as compared with normal NK cells. These microRNAs inhibited the growth of NKTCL cell lines in vitro. Nevertheless, the expression of miR-155 and miR-378 were upregulated[69]. miR-150 may impact the sensitivity of NKTCL cells to radiotherapy by inhibiting the PI3K/AKT/mTOR pathway and participate in the development of lymphoma[70].
miR-15a is downregulated in NKTCL cell lines and tumor tissues, which may predict poor prognosis and response to treatment[71]. Paik et al.[72] proved that the overexpression of miR-146a in NKTCL cells may inhibit NF-κB pathway and thus serve as a feasible tumor suppressor. In addition, Liang et al.[73] found that miR-223 targeted the 3’-untranslated region (UTR) of PRDM1 gene to downregulate its expression at the post-transcriptional level. miR-21 is overexpressed in NKTCL, acting as an oncogenic RNA which mediates apoptotic activity reduction through the abnormally dysregulated PTEN/AKT signaling pathway. Aberrant overexpression of miR-155 may induce activation of SHIP1/AKT pathway and affect the occurrence of NKTCL[74].
lncRNAs
In addition to microRNAs, some long non-coding RNAs (lncRNAs) have also been thought to be involved in the oncogenesis of NKTCL. ZFAS1 suggested genes participate in critical pathways associated with cell growth and tumor transformation, such as NF-κB and WNT signaling pathways[75]. Recently, Wang et al.[76] discovered that brain cytoplasmic RNA 1 in NKTCL tissue was significantly higher in contrast to normal NK cells, which may induce autophagy via inhibiting PI3K/AKT/mTOR and p53/mTOR signaling pathways and enhance the resistance to L-asparaginase. lncRNA X-inactive-specific transcript (XIST) is overexpressed in NKTCL, and its downstream target miR-497 could decrease the synthesis of the anti-apoptotic molecule Bcl-w to further regulate XIST-mediated proliferation and migration of NKTCL cells[77]. In addition to these, small nucleolar RNA host gene 12 is upregulated in NKTCL, as a direct transcription target of c-myc, which accelerates the proliferation of tumor cells and may suppress the response to cisplatin[78].
In summary, studies on the connection between non-coding RNAs and NKTCL not only present the possible therapeutic value of non-coding RNA but also provide enlightenment for overcoming the resistance of NKTCL patients. These potential targets and their significance are summarized in Table 4.
Important non-coding RNAs and their potential roles and therapeutic significance associated with NKTCL
| The alterations of human genetics | Potential hallmark/signaling pathways | Role in pathogenic mechanism of lymphoma | Ref. | Potential treatment significance | Ref. |
| microRNAs | miR-15a | Downregulated in NKTCL cell lines | Komabayashi et al.[71] | May predict poor response to treatment and survival of NKTCL patients | Komabayashi et al.[71] |
| miR-21 | Upregulated and mediates apoptotic activity reduction | Yamanaka et al.[74] | N/A | N/A | |
| miR-146a | Overexpressed and may limit Bcl-2 expression by inhibiting NF-κB pathway | Paik et al.[72] | N/A | N/A | |
| miR-150 | Downregulated in NKTCL tumor tissues and in vitro | Ng et al.[69] | Overexpression of miR-150 impacts the sensitivity to radiotherapy | Wu et al.[70] | |
| miR-155 | Overexpressed and induce aberrant activation of SHIP1/AKT pathway | Yamanaka et al.[74] | N/A | N/A | |
| miR-223 | Upregulated and targets and downregulates PRDM1 | Liang et al.[73] | N/A | N/A | |
| lncRNAs | BCYRN1 | Upregulated and may induce autophagy | Wang et al.[76] | Aberrant expression of BCYRN1 enhances resistance to L-asparaginase. | Wang et al.[76] |
| XIST | Overexpressed and mediates proliferation and migration in vitro | Liu et al.[77] | N/A | N/A | |
| SNHG12 | Upregulated and advances cell proliferation as a direct transcription target of c-myc | Zhu et al.[78] | C-myc may suppress the response to cisplatin by promoting SNHG12 expression in NKTCL | Zhu et al.[78] |
Immune-related changes
PD-1 and PD-L1
Programmed death-1 (PD-1) is an immune checkpoint receptor that binds to PD-L1 expressed by neoplasm cells and inhibits the activation, proliferation, and cytokine expression of T cells[79]. The combination of PD-1 and PD-L1 is one of the most important pathways for tumors to escape immune surveillance[80]. PD-1/PD-L1 blockade in various tumors, such as relapsed/refractory Hodgkin’s lymphoma[81], non-small-cell lung cancer[82], melanoma[83], and colorectal cancer[84], has been verified to be a newly favorable treatment option distinctive from traditional chemotherapeutics.
There are also studies on PD-1/PD-L1 blockade therapy applied to NKTCL patients. In the trial of
A meta-analysis including 4174 cases of five types of advanced or metastatic tumors displayed that PD-1/PD-L1 inhibitors are more effective than conventional chemotherapy, and the overall survival (OS) of patients is significantly prolonged, whether PD-L1 positive or negative[89]. Based on the above research reports, PD-1/PD-L1 blockade is a promising molecular-targeted approach. It is necessary to further explore how to improve the therapeutic effect and safety of PD-1/PD-L1 blockade therapy in NKTCL.
HLA risk alleles
A Japanese study containing 25 NKTCL cases and 303 control individuals reported that the frequency of human leukocyte antigen (HLA)-A*0201 in NKTCL patients was significantly lower than the baseline control population[90]. However, genome-wide association study did not observe similar results, probably due to the small sample size. Notably, Li et al.[15] identified 51 single-nucleotide polymorphisms (SNPs) associated with NKTCL that are mapped to the MHC region of chromosome 6. rs9277378 (located in HLA-DPB1) exhibits the strongest association with susceptibility of NKTCL [P = 4.21 × 10−19, odds ratio (OR) = 1.84]. Afterwards, Lin et al.[16] reported two novel NKTCL risk loci, the IL18RAP region on 2q12.1 (rs13015714; P = 2.83 × 10−16, OR = 1.39) and the HLA-DRB1 region on 6p21.3 (rs9271588; P = 9.35 × 10−26, OR = 1.53). The rs1420106-A variant that is highly correlated with rs13015714 can upregulate the expression of IL18RAP, which may be conducive to the proliferation of tumor cells. In addition, a haplotype association analysis showed that 47F-67I, a component of the antigen binding pocket of HLA-DRB1, was associated with a reduced risk of NKTCL, and 47Y-67L was the opposite to 47F-67I for the genetic risk for NKTCL[16].
Furthermore, a seven-SNP-based classifier was designed based on the seven SNPs that correlated to WDR27, UMAD1, TENM2, LINC02463, KDM4C, FGD4, and FAM71A. It had better predictive accuracy than clinicopathological risk variables on the survival of NKTCL patients. The combined application of the seven-SNP-based classifier and clinicopathological risk factor should be more accurate for predicting the prognosis of NKTCL patients[91].
All the above research provides new insights for the tumorigenesis and development of NKTCL and reveals the significance of inflammation and immune regulation through the IL18/IL-18RAP axis and antigen presentation involving HLA-DRB1. The relevant content is summarized in Table 5. The above implicated guiding significance for the risk stratification of NKTCL patients and clinical intervention. It could be combined with other genetic risk factors or prognostic models to help identify high-risk populations for targeted prevention.
Crucial immune checkpoint and risk alleles and their potential significance to the pathogenesis and treatment of NKTCL
| The alterations of human genetics | Potential hallmark/signaling pathways | Role in pathogenic mechanism of lymphoma | Ref. | Potential treatment significance | Ref. |
| Immune evasion | PD-1/PD-L1 | The combination of PD-1 and PD-L1 is one of the most important pathways for tumors to escape immune surveillance | Iwai et al.[80] | PD-1/PD-L1 blockade therapies applied to NKTCL have been developed or are ongoing, and there are a few promising results | Kwong et al.[85] Li et al.[86] Shen et al.[89] Lim et al.[87] Kim et al.[88] |
| HLA risk alleles | rs9277378 | Exhibits the strong association with susceptibility of NKTCL | Li et al.[15] | Susceptible population screening and disease risk stratification | N/A |
| rs13015714 | Located at the IL18RAP region and involved in inflammation and immune regulation | Lin et al.[16] | Identifying high risk population for targeted prevention | N/A | |
| rs9271588 | Located at the HLA-DRB1 region and involved in antigen presentation | Lin et al.[16] | Identifying the high-risk population for targeted prevention | N/A |
THE VARIATIONS OF EBV GENOME
Epstein-Barr virus (EBV) is a widespread human herpes virus that has infected more than 90% of population in a lifetime[92]. EBV infection is believed to be associated with various human cancers such as nasopharyngeal carcinoma, Burkitt’s lymphoma, Hodgkin’s lymphoma, gastric cancer, DLBCL, NKTCL,
Structural variation of EBV genome
There are common intragenic EBV deletions (73-49,847 bp) detected in NKTCL (10/23), EBV-positive DLBCL (10/14), and other malignancies (2/7)[98]. Sanger sequencing revealed that LMP1 gene contained a 30 bp deletion, which may be related to the poor prognosis of NKTCL patients[99]. Analyzing the EBV genome and transcriptome derived from NKTCL, in addition to the 30 bp deletion in LMP1, small deletions in BARTs, EBNA2, EBNA3s, BLLF1/2, and other regions were also detected, which disclosed the heterogeneity in EBV cloning in NKTCL patients[17]. Interestingly, this study also clarified an insertion of EBV fragments into the human nonhomologous end-joining 1 (NHEJ1) gene region, which may lead to changes in the expression and function of NHEJ1. The NHEJ1 gene is vital in repairing DNA damage and maintaining genome stability[100]. Thus, integration of the EBV genome and human genome might have crucial impact on the pathogenesis and development of NKTCL. The molecular mechanism of this integration affecting NKTCL tumorigenesis and whether the integration indeed contributes to clinical treatment of NKTCL need to be explored in further research.
Lytic genes
There are two infection routes of EBV-infected cells: latent infection and lytic infection[101]. Similar to latent genes, lytic genes also play a crucial role in the promotion of EBV infection and tumorigenesis of NKTCL. Previous studies demonstrated that the lytic genes BNLF2a and BNLF2b were highly expressed in NKTCL tissues[17], and the expressions of BARF1, BHRF1, and BZLF1 were detected in the NKTCL cell line[102]. Besides, the single-nucleotide variations of lytic gene BALF3 frequently were detected, and overexpression of BALF3 might drive DNA damage and bring about genomic instability in NKTCL. Intragenic EBV deletions often affect BamHI A rightward transcript (BART) microRNA clusters, core genes necessary for lytic DNA replication (BMRF1, BSLF1, BALF2, BALF5, BBLF2/BBLF3, and BBLF4) and some genes related to the lytic cycle and latent infection[98]. High-throughput sequence identified that EBV noncoding BART lncRNAs RPMS1 and A73 were strongly expressed in NKTCL and delivered regulatory signals to host cells without triggering specific immunity, which is helpful to retain the latent state of EBV in the host[103]. However, deletions located on (BART) microRNA clusters often have an unfavorable effect on EBV-miR-BART6-5p, EBV-miR-BART6-3p, EBV-miR-BART18-5p, and EBV-miR-BART20-5p, which negatively regulated early genes BZLF1 and BRLF1 that are thought to upregulate the lysis cycle and promote lymphoma pathogenesis[104-106].
Immune cell therapies targeted EBV
Studies have proved that EBV is closely related to the pathogenesis of NKTCL. While exploring the relevant molecular pathogenic mechanisms, scholars commit to explore the target of EBV for therapy. After 29 high-risk or recurrent cases with EBV+ lymphoma received autologous LMP-cytotoxic T lymphocytes (CTLs) therapy, 27 patients achieved CR[107]. Similarly, allogeneic donor-derived LMP-specific T cells (LMP-Ts), as an ancillary therapy, maintained the clinical response of EBV+ lymphoma patients who had underwent allogeneic bone marrow transplantation, and the two-year OS of 26 patients was 68%[108]. Noticeably, a recent study reported that EBV-specific induced pluripotent stem cell-derived antigen-specific CTLs can prompt enduring and strong antineoplastic activity[109]. The above research implies that specific immune cell therapies targeting EBV-related antigens are potential strategies. In the future, the safety and effectiveness of cell therapy should be further verified in larger multi-center trials which may bring more clinical benefits to NKTCL patients.
CONCLUSION
During the past decade, with the development of gene expression profiling and next-generation sequencing technology, people have expanded the understanding of the functional structure of genes. This provides novel perspectives for exploring the genetic mechanism of NKTCL and opportunities to develop new therapeutic strategies for NKTCL patients. Up to now, PD-1/PD-L1 blockade therapy has undoubtedly been an extremely noteworthy treatment option. As a novel regulator, CMTM6 regulates PD-L1 through endosystemic circulation, and PD-L1 can be specifically reduced due to the deletion of CMTM6. This discovery will be an interesting direction for further uncovering the NKTCL pathogenic mechanism[110]. Dysregulated signaling pathways are of noticeable significance in NKTCL and studies about related inhibitors also brings potential targets for the treatment of NKTCL[45,46,52,58,59]. The intimate relationship between EBV infection and NKTCL has been clearly proposed, and specific immune cell therapies targeting EBV are being investigated. Nevertheless, the interaction mechanism between the EBV genome and the human genome in NKTCL is yet to determined. As the results of various basic and clinical trials have been reported, feasible targets and possible treatments of NKTCL have been proposed and further studied. To sum up, more accurate biomarkers and predictors for the response to treatment and survival are needed. More effective treatment and the optimal combination of these therapeutic options still need to be explored in the future according to the development of the above progress and further challenges.
DECLARATIONS
Authors’ contributionsConception and design: Peng R, Jiang J
Acquisition of data: Jiang J, Ruan Z, Wang Q, Jiang L
Writing, review, and/or revision of the manuscript: Peng R, Jiang J
Administrative, technical, or material support: Ruan Z, Wang Q
Study supervision: Peng R
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work was supported by Guangzhou Science and Technology Plan Project (Grant No. 201804010024).
Conflicts of interestNot applicable.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Harabuchi Y, Yamanaka N, Kataura A, Imai S, Kinoshita T, Osato T. Epstein-Barr virus in nasal T-cell lymphomas in patients with lethal midline granuloma. Lancet 1990;335:128-30.
2. Aozasa K, Ohsawa M, Tajima K, et al. Nation-wide study of lethal mid-line granuloma in Japan: frequencies of wegener's granulomatosis, polymorphic reticulosis, malignant lymphoma and other related conditions. Int J Cancer 1989;44:63-6.
3. Gaal K, Sun NC, Hernandez AM, Arber DA. Sinonasal NK/T-cell lymphomas in the United States. Am J Surg Pathol 2000;24:1511-7.
4. Nagata H, Konno A, Kimura N, et al. Characterization of novel natural killer (NK)-cell and gammadelta T-cell lines established from primary lesions of nasal T/NK-cell lymphomas associated with the Epstein-Barr virus. Blood 2001;97:708-13.
5. Ko YH, Ree HJ, Kim WS, Choi WH, Moon WS, Kim SW. Clinicopathologic and genotypic study of extranodal nasal-type natural killer/T-cell lymphoma and natural killer precursor lymphoma among Koreans. Cancer 2000;89:2106-16.
6. Gualco G, Domeny-Duarte P, Chioato L, Barber G, Natkunam Y, Bacchi CE. Clinicopathologic and molecular features of 122 Brazilian cases of nodal and extranodal NK/T-cell lymphoma, nasal type, with EBV subtyping analysis. Am J Surg Pathol 2011;35:1195-203.
7. Kaur K, Kakkar A, Bhardwaj N, et al. Spectrum of cytomorphological features of extranodal NK/T-cell lymphoma, nasal type. Cytopathology 2019;30:393-401.
8. Huang Y, Chen S, Wei R, et al. CD20-positive extranodal NK/T cell lymphoma: clinicopathologic and prognostic features. Virchows Arch 2020;477:873-83.
9. Au WY, Weisenburger DD, Intragumtornchai T, et al. International Peripheral T-Cell Lymphoma Project. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood 2009;113:3931-7.
10. Nagato T, Ohkuri T, Ohara K, et al. Programmed death-ligand 1 and its soluble form are highly expressed in nasal natural killer/T-cell lymphoma: a potential rationale for immunotherapy. Cancer Immunol Immunother 2017;66:877-90.
12. Fox CP, Civallero M, Ko Y, et al. Survival outcomes of patients with extranodal natural-killer T-cell lymphoma: a prospective cohort study from the international T-cell Project. Lancet Haematol 2020;7:e284-94.
13. Huang Y, de Reyniès A, de Leval L, et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood 2010;115:1226-37.
14. Montes-Mojarro IA, Chen BJ, Ramirez-Ibarguen AF, et al. Mutational profile and EBV strains of extranodal NK/T-cell lymphoma, nasal type in Latin America. Mod Pathol 2020;33:781-91.
15. Li Z, Xia Y, Feng L, et al. Genetic risk of extranodal natural killer T-cell lymphoma: a genome-wide association study. Lancet Oncol 2016;17:1240-7.
16. Lin G, Xu C, Chen K, et al. Genetic risk of extranodal natural killer T-cell lymphoma: a genome-wide association study in multiple populations. Lancet Oncol 2020;21:306-16.
17. Peng RJ, Han BW, Cai QQ, et al. Genomic and transcriptomic landscapes of Epstein-Barr virus in extranodal natural killer T-cell lymphoma. Leukemia 2019;33:1451-62.
18. Xiong J, Cui BW, Wang N, et al. Genomic and transcriptomic characterization of natural killer T cell lymphoma. Cancer Cell 2020;37:403-419.e6.
19. Lee SH, Singh I, Tisdale S, Abdel-Wahab O, Leslie CS, Mayr C. Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 2018;561:127-31.
20. Weinhold N, Ashby C, Rasche L, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016;128:1735-44.
21. Dong G, Li Y, Lee L, et al. Genetic manipulation of primary human natural killer cells to investigate the functional and oncogenic roles of PRDM1. Haematologica 2021;106:2427-38.
22. Kallies A, Carotta S, Huntington ND, et al. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood 2011;117:1869-79.
23. Karube K, Nakagawa M, Tsuzuki S, et al. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood 2011;118:3195-204.
24. Küçük C, Iqbal J, Hu X, et al. PRDM1 is a tumor suppressor gene in natural killer cell malignancies. Proc Natl Acad Sci U S A 2011;108:20119-24.
25. Desmots F, Roussel M, Pangault C, et al. Pan-HDAC inhibitors restore PRDM1 response to IL21 in CREBBP-mutated follicular lymphoma. Clin Cancer Res 2019;25:735-46.
26. Kim BR, Park SH, Jeong YA, et al. RUNX3 enhances TRAIL-induced apoptosis by upregulating DR5 in colorectal cancer. Oncogene 2019;38:3903-18.
27. Haider A, Steininger A, Ullmann R, et al. Inactivation of RUNX3/p46 promotes cutaneous T-Cell lymphoma. J Invest Dermatol 2016;136:2287-96.
28. Chisholm KM, Bangs CD, Bacchi CE, Molina-Kirsch H, Cherry A, Natkunam Y. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294-303.
29. Ng SB, Selvarajan V, Huang G, et al. Activated oncogenic pathways and therapeutic targets in extranodal nasal-type NK/T cell lymphoma revealed by gene expression profiling. J Pathol 2011;223:496-510.
30. Selvarajan V, Osato M, Nah GSS, et al. RUNX3 is oncogenic in natural killer/T-cell lymphoma and is transcriptionally regulated by MYC. Leukemia 2017;31:2219-27.
32. Jin J, Wang J, Chen F, et al. Homoharringtonine-based induction regimens for patients with de-novo acute myeloid leukaemia: a multicentre, open-label, randomised, controlled phase 3 trial. Lancet Oncol 2013;14:599-608.
33. Yan J, Ng SB, Tay JL, et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood 2013;121:4512-20.
34. McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012;492:108-12.
35. Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010;42:181-5.
36. Yan J, Li B, Lin B, et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016;128:948-58.
37. Li B, Yan J, Phyu T, et al. MELK mediates the stability of EZH2 through site-specific phosphorylation in extranodal natural killer/T-cell lymphoma. Blood 2019;134:2046-58.
38. Italiano A, Soria J, Toulmonde M, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 2018;19:649-59.
39. Patmore DM, Jassim A, Nathan E, et al. DDX3X suppresses the susceptibility of hindbrain lineages to medulloblastoma. Dev Cell 2020;54:455-470.e5.
40. Phung B, Cieśla M, Sanna A, et al. The X-Linked DDX3X RNA helicase dictates translation reprogramming and metastasis in melanoma. Cell Rep 2019;27:3573-3586.e7.
41. Dobashi A, Tsuyama N, Asaka R, et al. Frequent BCOR aberrations in extranodal NK/T-Cell lymphoma, nasal type. Genes Chromosomes Cancer 2016;55:460-71.
42. Jiang L, Gu ZH, Yan ZX, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet 2015;47:1061-6.
43. Choi S, Go JH, Kim EK, et al. Mutational analysis of extranodal NK/T-Cell lymphoma using targeted sequencing with a comprehensive cancer panel. Genomics Inform 2016;14:78-84.
44. Chen Q, Feng J, Wu J, et al. HKDC1 C-terminal based peptides inhibit extranodal natural killer/T-cell lymphoma by modulation of mitochondrial function and EBV suppression. Leukemia 2020;34:2736-48.
45. Song TL, Nairismägi ML, Laurensia Y, et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018;132:1146-58.
46. Lee S, Park HY, Kang SY, et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget 2015;6:17764-76.
47. Coppo P, Gouilleux-Gruart V, Huang Y, et al. STAT3 transcription factor is constitutively activated and is oncogenic in nasal-type NK/T-cell lymphoma. Leukemia 2009;23:1667-78.
48. Chen YW, Guo T, Shen L, et al. Receptor-type tyrosine-protein phosphatase κ directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood 2015;125:1589-600.
49. Bouchekioua A, Scourzic L, de Wever O, et al. JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal nasal-type natural killer cell lymphoma. Leukemia 2014;28:338-48.
50. Koo GC, Tan SY, Tang T, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov 2012;2:591-7.
51. Sim SH, Kim S, Kim TM, et al. Novel JAK3-activating mutations in extranodal NK/T-cell lymphoma, nasal type. Am J Pathol 2017;187:980-6.
52. Nairismägi M-, Gerritsen ME, Li ZM, et al. Oncogenic activation of JAK3-STAT signaling confers clinical sensitivity to PRN371, a novel selective and potent JAK3 inhibitor, in natural killer/T-cell lymphoma. Leukemia 2018;32:1147-56.
53. Jost PJ, Ruland J. Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood 2007;109:2700-7.
54. Sun L, Zhao Y, Shi H, Ma C, Wei L. LMP1 promotes nasal NK/T-cell lymphoma cell function by eIF4E via NF-κB pathway. Oncol Rep 2015;34:3264-71.
55. Sun L, Zhao Y, Shi H, Ma C, Wei L. LMP-1 induces survivin expression to inhibit cell apoptosis through the NF-κB and PI3K/Akt signaling pathways in nasal NK/T-cell lymphoma. Oncol Rep 2015;33:2253-60.
56. Bi XW, Wang H, Zhang WW, et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol 2016;9:109.
57. Piccaluga PP, Rossi M, Agostinelli C, et al. Platelet-derived growth factor alpha mediates the proliferation of peripheral T-cell lymphoma cells via an autocrine regulatory pathway. Leukemia 2014;28:1687-97.
58. Iqbal J, Weisenburger DD, Chowdhury A, et al. International Peripheral T-cell Lymphoma Project. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic γδ T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro. Leukemia 2011;25:348-58.
59. Kawada J, Ito Y, Iwata S, et al. mTOR inhibitors induce cell-cycle arrest and inhibit tumor growth in Epstein-Barr virus-associated T and natural killer cell lymphoma cells. Clin Cancer Res 2014;20:5412-22.
60. Huang D, Song TL, Nairismägi ML, et al. Evaluation of the PIK3 pathway in peripheral T-cell lymphoma and NK/T-cell lymphoma. Br J Haematol 2020;189:731-44.
61. Lee WJ, Kim YJ, Lee YJ, et al. Vascular endothelial growth factor protein expression is associated with a poor prognosis in patients with cutaneous extranodal natural killer/T-cell lymphoma. Br J Dermatol 2018;178:e11-2.
62. Fearnley GW, Young KA, Edgar JR, et al. The homophilic receptor PTPRK selectively dephosphorylates multiple junctional regulators to promote cell-cell adhesion. Elife 2019;8:e44597.
63. Zhang L, Anglesio MS, O'Sullivan M, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med 2007;13:1060-9.
64. Sako N, Dessirier V, Bagot M, Bensussan A, Schmitt C. HACE1, a potential tumor suppressor gene on 6q21, is not involved in extranodal natural killer/T-cell lymphoma pathophysiology. Am J Pathol 2014;184:2899-907.
65. Cimmino L, Dawlaty MM, Ndiaye-Lobry D, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol 2015;16:653-62.
66. Poole CJ, Lodh A, Choi JH, van Riggelen J. MYC deregulates TET1 and TET2 expression to control global DNA (hydroxy)methylation and gene expression to maintain a neoplastic phenotype in T-ALL. Epigenetics Chromatin 2019;12:41.
67. Li L, Li C, Mao H, et al. Epigenetic inactivation of the CpG demethylase TET1 as a DNA methylation feedback loop in human cancers. Sci Rep 2016;6:26591.
68. Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med 2006;12:580-7.
69. Ng SB, Yan J, Huang G, et al. Dysregulated microRNAs affect pathways and targets of biologic relevance in nasal-type natural killer/T-cell lymphoma. Blood 2011;118:4919-29.
70. Wu SJ, Chen J, Wu B, Wang YJ, Guo KY. MicroRNA-150 enhances radiosensitivity by inhibiting the AKT pathway in NK/T cell lymphoma. J Exp Clin Cancer Res 2018;37:18.
71. Komabayashi Y, Kishibe K, Nagato T, Ueda S, Takahara M, Harabuchi Y. Downregulation of miR-15a due to LMP1 promotes cell proliferation and predicts poor prognosis in nasal NK/T-cell lymphoma. Am J Hematol 2014;89:25-33.
72. Paik JH, Jang JY, Jeon YK, et al. MicroRNA-146a downregulates NFκB activity via targeting TRAF6 and functions as a tumor suppressor having strong prognostic implications in NK/T cell lymphoma. Clin Cancer Res 2011;17:4761-71.
73. Liang L, Nong L, Zhang S, et al. The downregulation of PRDM1/Blimp-1 is associated with aberrant expression of miR-223 in extranodal NK/T-cell lymphoma, nasal type. J Exp Clin Cancer Res 2014;33:7.
74. Yamanaka Y, Tagawa H, Takahashi N, et al. Aberrant overexpression of microRNAs activate AKT signaling via down-regulation of tumor suppressors in natural killer-cell lymphoma/leukemia. Blood 2009;114:3265-75.
75. Baytak E, Gong Q, Akman B, Yuan H, Chan WC, Küçük C. Whole transcriptome analysis reveals dysregulated oncogenic lncRNAs in natural killer/T-cell lymphoma and establishes MIR155HG as a target of PRDM1. Tumour Biol 2017;39:1010428317701648.
76. Wang L, Yang J, Wang HN, et al. LncRNA BCYRN1-induced autophagy enhances asparaginase resistance in extranodal NK/T-cell lymphoma. Theranostics 2021;11:925-40.
77. Liu Q, Ran R, Wu Z, et al. Long Non-coding RNA X-inactive specific transcript mediates cell proliferation and intrusion by modulating the miR-497/Bcl-w axis in extranodal natural killer/T-cell lymphoma. Front Cell Dev Biol 2020;8:599070.
78. Zhu L, Zhang X, Fu X, et al. c-Myc mediated upregulation of long noncoding RNA SNHG12 regulates proliferation and drug sensitivity in natural killer/T-cell lymphoma. J Cell Biochem 2019;120:12628-37.
79. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677-704.
80. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002;99:12293-7.
81. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015;372:311-9.
82. Herbst RS, Baas P, Kim D, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387:1540-50.
83. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23-34.
84. André T, Shiu KK, Kim TW, et al. KEYNOTE-177 Investigators. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med 2020;383:2207-18.
85. Kwong YL, Chan TSY, Tan D, et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017;129:2437-42.
86. Li X, Cheng Y, Zhang M, et al. Activity of pembrolizumab in relapsed/refractory NK/T-cell lymphoma. J Hematol Oncol 2018;11:15.
87. Lim JQ, Huang D, Tang T, et al. Whole-genome sequencing identifies responders to Pembrolizumab in relapse/refractory natural-killer/T cell lymphoma. Leukemia 2020;34:3413-9.
88. Kim SJ, Lim JQ, Laurensia Y, et al. Avelumab for the treatment of relapsed or refractory extranodal NK/T-cell lymphoma: an open-label phase 2 study. Blood 2020;136:2754-63.
89. Shen X, Zhao B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: meta-analysis. BMJ 2018;362:k3529.
90. Kanno H, Kojya S, Li T, Ohsawa M, Nakatsuka S, et al. Low frequency of HLA-A*0201 allele in patients with Epstein-Barr virus-positive nasal lymphomas with polymorphic reticulosis morphology. Int J Cancer 2000;87:195-9.
91. Tian XP, Ma SY, Young KH, et al. A composite single-nucleotide polymorphism prediction signature for extranodal natural killer/T-cell lymphoma. Blood ;2021:452-63.
93. Young LS, Yap LF, Murray PG. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer 2016;16:789-802.
94. Chiang AK, Tao Q, Srivastava G, Ho FC. Nasal NK- and T-cell lymphomas share the same type of Epstein-Barr virus latency as nasopharyngeal carcinoma and Hodgkin's disease. Int J Cancer 1996;68:285-90.
95. Xu ZG, Iwatsuki K, Oyama N, et al. The latency pattern of Epstein-Barr virus infection and viral IL-10 expression in cutaneous natural killer/T-cell lymphomas. Br J Cancer 2001;84:920-5.
97. Humme S, Reisbach G, Feederle R, et al. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc Natl Acad Sci U S A 2003;100:10989-94.
98. Okuno Y, Murata T, Sato Y, et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat Microbiol 2019;4:404-13.
99. Nagamine M, Takahara M, Kishibe K, et al. Sequence variations of Epstein-Barr virus LMP1 gene in nasal NK/T-cell lymphoma. Virus Genes 2007;34:47-54.
100. Menon V, Povirk LF. XLF/Cernunnos: An important but puzzling participant in the nonhomologous end joining DNA repair pathway. DNA Repair (Amst) 2017;58:29-37.
101. Bauer CC, Aberle SW, Popow-Kraupp T, Kapitan M, Hofmann H, Puchhammer-Stöckl E. Serum Epstein-Barr virus DNA load in primary Epstein-Barr virus infection. J Med Virol 2005;75:54-8.
102. Zhang Y, Ohyashiki JH, Takaku T, Shimizu N, Ohyashiki K. Transcriptional profiling of Epstein-Barr virus (EBV) genes and host cellular genes in nasal NK/T-cell lymphoma and chronic active EBV infection. Br J Cancer 2006;94:599-608.
103. Nakhoul H, Lin Z, Wang X, Roberts C, Dong Y, Flemington E. High-throughput sequence analysis of peripheral T-Cell lymphomas indicates subtype-specific viral gene expression patterns and immune cell microenvironments. mSphere 2019;4:e00248-19.
104. Qiu J, Thorley-Lawson DA. EBV microRNA BART 18-5p targets MAP3K2 to facilitate persistence in vivo by inhibiting viral replication in B cells. Proc Natl Acad Sci U S A 2014;111:11157-62.
105. Iizasa H, Wulff BE, Alla NR, et al. Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J Biol Chem 2010;285:33358-70.
106. Jung YJ, Choi H, Kim H, Lee SK. MicroRNA miR-BART20-5p stabilizes Epstein-Barr virus latency by directly targeting BZLF1 and BRLF1. J Virol 2014;88:9027-37.
107. Bollard CM, Gottschalk S, Torrano V, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol 2014;32:798-808.
108. McLaughlin LP, Rouce R, Gottschalk S, et al. EBV/LMP-specific T cells maintain remissions of T- and B-cell EBV lymphomas after allogeneic bone marrow transplantation. Blood 2018;132:2351-61.
109. Ando M, Ando J, Yamazaki S, et al. Long-term eradication of extranodal natural killer/T-cell lymphoma, nasal type, by induced pluripotent stem cell-derived Epstein-Barr virus-specific rejuvenated T cells in vivo. Haematologica 2020;105:796-807.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Jiang J, Ruan Z, Wang Q, Jiang L, Peng R. Genetics and genomics of extranodal natural killer/T cell lymphoma: from etiology to treatment. J Transl Genet Genom 2021;5:323-40. http://dx.doi.org/10.20517/jtgg.2021.21
AMA Style
Jiang J, Ruan Z, Wang Q, Jiang L, Peng R. Genetics and genomics of extranodal natural killer/T cell lymphoma: from etiology to treatment. Journal of Translational Genetics and Genomics. 2021; 5(3): 323-40. http://dx.doi.org/10.20517/jtgg.2021.21
Chicago/Turabian Style
Jiang, Jiaxin, Zhaohui Ruan, Qianyu Wang, Li Jiang, Roujun Peng. 2021. "Genetics and genomics of extranodal natural killer/T cell lymphoma: from etiology to treatment" Journal of Translational Genetics and Genomics. 5, no.3: 323-40. http://dx.doi.org/10.20517/jtgg.2021.21
ACS Style
Jiang, J.; Ruan Z.; Wang Q.; Jiang L.; Peng R. Genetics and genomics of extranodal natural killer/T cell lymphoma: from etiology to treatment. J. Transl. Genet. Genom. 2021, 5, 323-40. http://dx.doi.org/10.20517/jtgg.2021.21
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 0 clicks
Cite This Article 0 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.