
Epigenetics in prostate cancer treatment


Abstract
Prostate cancer (PCa) is the most commonly diagnosed malignancy among men, and the progression of this disease results in fewer treatment options available to clinical patients. It highlights the vital necessity for discovering novel therapeutic approaches and expanding the current understanding of molecular mechanisms. Epigenetic alternations such as DNA methylation models and histone modifications have been associated as key drivers in the development and advancement of PCa. Several studies have been conducted and demonstrated that targeting these epigenetic enzymes or regulatory proteins has been strongly associated with the regulation of cancer cell growth. Due to the success rate of these therapeutic routes in pre-clinical settings, many drugs have now advanced to clinical testing, where efficacy will be measured. This review will discuss the role of epigenetic modifications in PCa development and its function in the progression of the disease to resistant forms and introduce therapeutic strategies that have demonstrated successful results as PCa treatment.
Keywords
INTRODUCTION
Prostate cancer (PCa) continues to be the most common cancer[1] and ranks as the second leading cause of cancer-related death in United States of America males[1]. Numerous studies have documented that androgen receptor (AR) signaling continues to participate as a vital component to the development and progression of PCa. AR is a nuclear hormone receptor that becomes activated upon the binding of androgen ligands and dissociates from cytoplasmic chaperone protein HSP90, where it then can self-dimerize and translocate to the nucleus[2]. AR has the ability to bind to androgen response elements (ARE), which results in the transcription of target genes and contribution to prostate development and maintenance[2]. In early diagnosed stages of the disease, there are successful treatment options to prolong patient survival, such as medical or surgical castration that distribute AR binding, including radical prostatectomy, radiotherapy, or androgen deprivation therapy (ADT). However, PCa can reoccur after these interventions, which is referenced as castration-resistant PCa (CRPC).
CRPC is defined as cancer that continues to proliferate in the absence or depletion of testosterone, compared to early stages that require higher levels of testosterone for survival and formation, but AR signaling remains an essential contributor to PCa progression. This led to the synthesis of Androgen-Signaling Inhibitors (ASI), such as Abiraterone and Enzalutamide (ENZ), which targets androgen synthesis and AR, respectively. Though ASI treatment options have shown impressive results in reducing PCa, enviably, within several months, the disease will progress into terminal ASI-resistant PCa. Currently, there are several various areas of study arising to address the issue of overcoming ASI resistance. Abiraterone acetate is a hormonal treatment for metastatic CRPC (mCRPC) both before and after chemotherapy and significantly reduces androgen production by blocking the enzyme, which illustrates its efficacy[2]. ENZ is a second-generation nonsteroidal antiandrogen utilized in the treatment of mCRPC patients and has shown efficacy to provide patients with a reasonable quality of life[3]. Though there has been significant success associated with the administration of these treatments, resistance does occur with both Abiraterone and ENZ, possibly from the development of splice variants[3]. Wnt signaling has been extensively documented in its involvement in ENZ resistance. The inhibition of both canonical and non-canonical Wnt signaling has demonstrated positive results in re-establishing sensitivity to ENZ treatment in resistant cell lines and tumor models[3-5].
Additionally, we have established that 3-hydroxy-3-methyl-glutaryl-CoA reductase, a crucial enzyme in the mevalonate pathway, is elevated in ENZ-resistant PCa cells, and combination therapy of simvastatin and ENZ could result in significant inhibition of ENZ-resistant cancer cell proliferation in both in vivo and in vitro models[6]. Even in terms of AR splice variants, which play a critical role in the development and progression of CRPC[7]. Wang et al.[8] demonstrated that treatment with Malat1 small interfering RNA inhibits ARV7 expression in cell lines and significantly reduces tumor growth in ENZ-resistant xenografts. Additionally, Yamamoto et al.[9] demonstrated that using antisense oligonucleotides to target both the full-length AR and its splice variants resulted in a suppression of ENZ-resistant cell lines and xenografts proliferation. Due to the increase in understanding the molecular mechanisms that drive PCa, there has been a rising interest in epigenetics related to new therapeutic approaches.
Epigenetics is the study of heritable changes in gene expression, subsequently controlling fate, without affecting actual DNA sequences[10]. An abundance of exploration has been accomplished in epigenetics since it was first introduced and defined in the 1950s by Conrad Waddington. These recent discoveries have led to the emergence of operational mechanisms that are composed of three processing steps. These steps can be defined as epigenator, which is a signal that originates from the cellular environment and initiates downstream signaling pathways[11]. The epigenetic initiator, which triggers the epigenetic modification at a specific chromatin structure and the epigenetic maintainers that are epigenetic code responsible for ensuring several events such as chemical modifications on DNA or histones molecules, interacts between DNA, RNA, non-coding RNAs, protein, and other chromatin remodeling events[11]. Regarding the study of PCa, these aberrant alternations have reinforced the establishment of a context-specific translational profile that favors self-renewal, survival, and invasion and has demonstrated that the accumulation of epigenetic aberrations eventually causes genetic or genomic instability [Figure 1]. Additionally, AR is demonstrated to function in conjunction with various chromatin remodelers and epigenetic players that regulate prostate development and its progression to a malignant phenotype. This review will discuss the essential epigenetic alternations that are critical in comprehending PCa etiology and developments that highlight new biomarkers and therapeutic approaches to PCa.
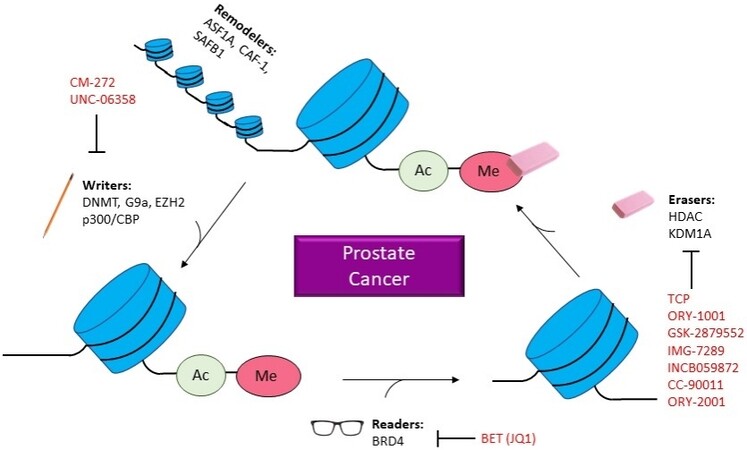
Figure 1. Epigenetics in PCa. Diagram illustrating how epigenetic changes related to prostate cancer and which inhibitors are in current studies. ASF1A: Anti-silencing function 1A hisotne chaperone; CAF-1: chromatin assembly factor 1; SAFB1: scaffold attachment factor B; DNMT: DNA methyltransferase; EZH2: enhancer of zeste 2 polycomb repressive complex 2 subunit; CBP: CREB-binding protein; BRD4: bromodomain containing 4; BET: bromodomain and extra terminal domain; HDAC: histone deacetylases; KDM1A: lysine demethylase 1A; CM-272: 6-methoxy-2-(5-methylfuran-2-yl)-N-(1-methylpiperidin-4-yl)-7-(3-(pyrrolidin-1-yl)propoxy)quinolin-4-amine; UNC-06358: 2-cyclohexyl-6-methoxy-N-(1-propan-2-ylpiperidin-4-yl)-7-(3-pyrrolidin-1-ylpropoxy)quinazolin-4-amine; JQ1: (S)-tert-butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate; TCP: tranylcypromine; ORY-1001: idademstat; IMG-7289: bomedemstat HCL; INCB059872: 1-((4-(methoxymethyl)-4-((((1R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin-1-yl)methyl)cyclobutane-1-carboxylic acid compound with 4-methylbenzenesulfonic acid (1:2); CC-90011: besylate; ORY-2001: vafidemstat.
EPIGENETIC REGULATORS OF PCa
Epigenetic writers
Epigenetic codes have commonly been documented to be regulated by writers, readers, and erasers. Writers hold the responsibility to transcribe the epigenetic modifications of DNA and histone proteins[11]. These modifications transpire from the addition of various chemical groups utilizing numerous enzymes. An invariable number of modifications have the potential to materialize, but for this review, we will focus on the reactions of methylation and acetylation. DNA and histone proteins are highly prone to methylation, which is the addition of a methyl group to a DNA molecule that may result in a change in the activity of the DNA segment, but will not modify the sequence. Routinely, acetylation is a process in which an acetyl functional group is transferred from molecule to an adjacent molecule and functions by removing the positive charge, thus reducing the N-termini interaction that contains negatively charged phosphates of DNA, exclusively in histones. In this area, we will focus on the addition of these modifications and how they affect the progression and severity of PCa. We will also explore therapeutic methodologies that have been established to address these alterations in function.
DNMT and DNA methylation
DNA methylation often plays a role in suppressing gene transcription when located in a gene promoter. DNA methyltransferases (DNMTs) are responsible for transferring methyl groups from the methyl donor S-adenosyl-L-methionine to the 5-position of cytosine residues in DNA, which is critical for mammalian development. The DNMT family has five members, including DNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3l[12]. DNMTs play an important role in genome integrity as their disruption may lead to chromosomal instability and tumor progression[12,13]. The main function of DNMT1 is to maintain the methylation status of DNA. As an RNA methyltransferase, DNMT2 usually methylates multiple tRNAs[14]. DNMT3a and DNMT3b are reported to contribute the de novo DNA methylation.
DNMT3l improves the catalytic activities of DNMT3a and DNMT3b, resulting in the promotion of DNA de novo methylation by interacting with DNMT3a and DNMT3b[15]. DNA methylation has been shown to play a role in PCa, and DNA methylation marks have been studied for their diagnostic and prognostic values. One of the most recognized DNA methylation events in prostate carcinogenesis is the hypermethylation of the regulatory region of GSTP1, leading to a decrease in gene expression. This hypermethylation event has been found in more than 90% of prostate adenocarcinoma samples and studied for its potential diagnostic and prognostic value[16]. Other studies have shown that various genes such as APC, RASSF1a, PTGS2, MDR1, GSTM2, and PENK are hypermethylated in primary and metastatic PCa cells compared to normal prostatic tissues, suggesting that DNA methylation becomes deregulated and may play a role in the prostate carcinogenesis process[16]. Both DNA hypermethylation and DNA hypomethylation correlate with prostate carcinogenesis and progression. In a study of 10 normal prostates and 45 prostate tumors, 61 genes were found to be hypermethylated in more than 20% of tumors. A cluster of tumors with hypermethylation of ETV1 and ZNF215 was correlated with ADT resistance in these patients, suggesting a potential use for hypermethylation cluster for prognostic purposes[17]. In another study analyzing 84 prostatic tumor tissues with low and intermediate grade PCa, DNA hypermethylation was associated with poorer prognosis and prostate-specific antigen (PSA) recurrence following prostatectomy[18]. Comparing DNA methylation in PCa tissues to benign prostatic hyperplasia tissues revealed a higher occurrence of hypermethylation in a group of genes, suggesting a role for these signatures in the diagnostic and prognostic setting of PCa[18]. The hypomethylation of MYC’s exon3 is not associated with changes in its expression; however, it was associated with a more aggressive phenotype in the examined cohort[19]. In another study and contrary to the established consensus, a group of hypermethylated genes in PCa tumors was associated with increased gene expression[20]. In an Iranian study, 35 prostate tumor samples were examined before and after hormone therapy treatment. Treatment with bicalutamide-based drugs for three months induced a significant decrease in the expression of DNMT3A and significant increases in the expression of DNMT3B and two well-established methylated genes, GSTP1 and RASSF1[21]. Gravina et al.[22] showed that treatment with bicalutamide induced an increase in DNMT activity in PCa that correlated with an increased expression of DNMT3A and DNMT3B. These observations warrant further investigation to understand better the regulation of DNA methylation patterns in PCa, their effects on disease progression, and treatment decisions moving forward.
G9a and histone Methylation
G9a, a histone methyltransferase, has the capacity to di-methylate histone 3 at lysine position 9. This epigenetic modification generally represses gene expression[23]. However, several studies have demonstrated that G9a also functions as a coactivator of nuclear receptors, such as AR[24,25]. Despite the epigenetic function of G9a, the first example of G9A operating as a non-histone lysine methyltransferase, reported that G9A could auto-methylate at the end of its N-terminal[26]. Following this trend, CDYL1, WIZ, and ACINUS were discovered as G9A substrates via peptide arrays[27], suggesting that G9A mediated lysine methylation is critical for both histone and non-histone proteins. It has been observed that G9a is overexpressed in a number of cancers[28,29], and elevated G9A protein and its enzymatic activities have been determined under hypoxia stress. For example, Reptin and Pontin, two chromatin remodeling factors, can be methylated by G9A through hypoxia-dependent manners[30,31]. Furthermore, methylated Retin contributes to tumor growth and invasive activities via negative regulation of HIF1. Additionally, hypoxia-induced Pontin methylation enhances the ability of proliferation and invasion in breast cancer cells. An alternative study showed that hypoxia-mediated G9A also suppressed RUNX, a tumor suppressor, through histone modification. In addition, hypoxia-mediated G9A amplification decreases apoptosis and increases immature stem-like cancer cells[32]. In contrast, hypoxia-mediated G9A represses cell adhesion molecules and contributes to breast tumor motility[33]. Additionally, G9A can also promote breast cancer cell survival through driving hypoxia-mediated gene expression. These impacts on cell malignant behavior potentially are caused by FIH-mediated G9A/GLP hydroxylation[34]. In addition, G9a also has been found to contribute to the aberrant metabolism of cancer cells. Increased G9a can epigenetically activate the serine biosynthesis, which in turn promotes cancer cell proliferation and survival[35]. Also, Fructose-1,6-bisphosphatase (FBP1), a rate-limiting enzyme, can catalyze F-1,6-BP into fructose 6-phosphate in gluconeogenesis. This process can be repressed by G9A mediated epigenetic modification in breast cancer cell lines. Repressed FBP1 contributes to epithelial-mesenchymal transition transformation, promoting cancer cell metastasis[36]. Loss of G9A initiates HEPH expression that converts Fe2+ into Fe3+. Excessive Fe3+ will initiate cell cycle arrest machinery[37]. Though, there is a lack of evidence that suggests that dysregulation of G9a affects PCa. The interaction between G9a and NKX3.1 contributes to prostate differentiation[38]. In addition, G9a plays as a coactivator for PSA induction[23]. It suggests that misregulation of G9a may possibly contribute to the generation and progression of PCa. In conclusion, inhibition of G9a may enhance cancer treatment, making it a promising target. The inhibition of G9a has been studied in various cancer types. CM-272, an inhibitor for both G9a and DNMTs, activates immune-related pathway and increase the efficacy of anti-PD-1 immunotherapy
Inhibitors for epigenetic changes
| Inhibitor | Target | Clinical trial phase |
| CM-272 | G9a | Not in clinical trial |
| UNC-0638 | G9a | Not in clinical trial |
| TCP | KDM1A | Phase 1/2 |
| ORY-1001 | KDM1A | Phase 1 |
| GSK-2879552 | KDM1A | Phase 1/2 |
| IMG-7289 | KDM1A | Phase 2 |
| INCB059872 | KDM1A | Phase 1/2 |
| CC-90011 | KDM1A | Phase 1 |
| ORY-2001 | KDM1A | Phase 2 |
| BET (JQ1) | BRD4 | Phase 1 |
EZH2 and histone methylation
Enhancer of zeste homolog 2 (EZH2) is the essential subunit of the polycomb repressor complex2 (PRC2) and acts as a histone methyltransferase to catalyze tri-methylation of Lys27 on histone H3 (H3K27me3). EZH2 is commonly known to promote the progression of diverse human cancers by H3K27me3-mediated silencing of tumor suppressors[41,42]. However, EZH2 can also methylate target genes directly, such as STAT3, GATA4, and Jarid2, to modulate their expression and contribute to cancer development[43-45]. In addition to the catalytic function of EZH2 in epigenetic modification, a novel PRC2-independent role of EZH2 as a transcriptional activator was identified by several studies, including NOTCH1, NF-κB, and Wnt signaling[46-48]. In the development of PCa, specifically CRPC, EZH2 has been identified to function as a transcriptional coactivator interacting with AR. This functional transfer from a transcriptional suppressor to an activator is driven by the AKT-dependent phosphorylation of EZH2 at Serine-21[49]. Recently, it was documented that EZH2 can activate AR signaling via direct binding at the AR promoter region[50]. According to these established molecular mechanisms contributing to ADT-resistance acquisition, our lab has questioned whether EZH2 contributes to the resistance of ENZ in CRPC. Our lab determined that EZH2 can bind to the promoter of PSA, resulting in the suppression of its transcription, concluding that pharmaceutical inhibition of EZH2 can overcome ENZ-resistance in CRPC[51]. Our findings suggest that the inhibition of EZH2 via existing FDA-approved EZH2 inhibitors can increase the efficacy of ENZ treatment, providing terminal CRPC patients with a novel therapeutic strategy. In addition, we also illustrated EZH2 inhibition could enhance the anti-neoplastic activity of metformin in PCa by reducing the binding of AR to the miR-26a-5p promoter[52]. Collectively, these findings suggest that EZH2 could be an effective therapeutic target for PCa, particularly for AR-positive CRPC.
p300/CBP and histone acetylation
Histone acetyltransferase p300 and its highly homologous CREB-binding protein (CBP) attach acetyl groups to proteins, including histones, in which DNA is wrapped[53,54]. Histone acetylation is a critical method that governs chromatin. When histones are acetylated, chromatin structures in that region will gain a loose conformation, and gene transcription will be promoted[54]. It has been reported that p300 and CBP were implicated in the progression of PCa and that deletion of p300 in mice limited PCa progression and extended mice survival[55]. The oncogenic roles of p300/CBP in the progression of PCa are usually related to the regulation of AR, the key driver of PCa. p300 can directly acetylate AR, or bind with AR, to enhance its transcriptional activity, consequently inducing oncogenes expression and promoting tumor growth[55-57]. In addition to enhancing AR transcriptional activity, p300 can also regulate AR protein level by preventing its degradation[55]. These findings highlight p300 as a compelling target for PCa treatment. Indeed, studies have shown that targeting p300/CBP inhibited both androgen-sensitive and CRPC cell growth[53,57,58]. In addition, our lab has recently reported a novel mechanism underlying p300 involvement in PCa progression by upregulating PD-L1 expression, thus creating an immune cell-free microenvironment for tumor progression. We found that p300 was recruited to the promoter of CD274 (encoding PD-L1) by transcription factor IRF-1 and resulted in acetylation of histone H3 at the CD274 promoter, and subsequently CD274 transcription. The p300/CBP inhibitor blocked the transcription of CD274 and hindered exosomal PD-L1 secretion. Cutting off PD-L1 secretion at transcription by inhibiting p300/CBP in combination with anti-PD-L1 antibodies demonstrated increased efficacy in a syngeneic mouse model of PCa[59]. Our discovery suggests that p300 is not only a modifier but also a co-driver for PCa progression, confirming that p300 could be a compelling target for PCa treatment.
Epigenetic readers
The framework of modifications constructed by epigenetic writers requires other cellular proteins to both recognize and mediate their effects. Epigenetic readers are protein domains that can bind to these modifications that may be present on DNA and histones. This section will focus on the domains that can be both and recognize methylation and acetylation and therapeutic approaches in PCa.
Readers of DNA methylation
DNA methylation is a major epigenetic process that regulates chromatin structure which causes transcriptional activation or repression of genes. The process of DNA methylation is the addition of methyl groups to the correct bases located on the genome by “writer” molecules, known as DNA methyltransferases[17]. DNA methylation can provide two different functions. The first function is that DNA methylation directly inhibits transcription factor binding at the gene regulatory region, resulting in transcriptional repression. An alternative operational route is to recruit reader molecules, commonly referred to as methyl-binding proteins (MBP), at the methylated site, which can then attract various members of the chromatin remodeling complex, which will result in transcriptional activation or repression with a dependence on the cellular content. DNA methylation has long been suspected of playing a role in tumorigenesis and cancer progression in various tissue types. Due to this linkage, several drugs have been approved by the FDA, such as Vidaza and Dacogen, which act as DNA methylation inhibitors and are utilized as cancer therapies. These inhibitors operate by reversing the hypermethylated state at the promoter regions of tumor suppressor genes and induce activation of premetastatic genes. In prostate cancer, it has been reported that the knockdown of methyl-binding protein 1 (MBP-1), which functions as a general transcriptional repressor in human PCa cells, results in a delay of cell cycle progression via the inhibition of cyclin A and cyclin B1 expression[60]. Additionally, it has been shown that the carboxyl-terminal repressor domain of MBP-1 (MBP-CR) is sufficient for regression of prostate tumor growth in nude mice and suggests that MBP-CR expression has an anti-proliferative effect in human prostate cancer cells compared to the full-length MBP-1 in preventing tumor growth[60].
BRD4
The bromodomain-containing family proteins recognize and bind to acetylated lysine residue modifications of histones or proteins, an important class of acetylation readers. The bromodomain was first reported as an evolutionarily conserved domain in proteins of humans, flies, and yeast in 1992[61]. It has approximately 110 amino acids and consists of four α helices forming a hydrophobic cavity that identifies acetyl-lysine. There are 42 bromodomain-containing proteins with 61 unique bromodomains In humans, in which differences of the amino acid residues at the acetyl-lysine binding site determine the specificity of binding[62]. The BET (bromodomain and extra terminal domain) subfamily proteins have two conserved amino-terminal bromodomains (BD1 and BD2) that are pivotal for recognizing acetylated lysine residues of histones and other non-histone proteins, playing an important role in regulating transcription by recruiting RNA polymerase II (POL II)[63]. BRD4 is one of the well-studied BET family proteins that recognize either histone tail or non-histone acetylated modifications at lysine residues. BRD4 was first described as a MED1-interacting protein and occupies thousands of enhancers and promoters related to gene activation[64]. BRD4 also works as a critical regulator of the positive transcriptional elongation factor b (P-TEFb) complex via recruiting it to the chromatin and mediates the activation of P-TEFb, consequently phosphorylating and activating RNAPOL II. It is reported that the interaction of BRD4/P-TEFb is crucial for rapid transcriptional reinitiating after mitosis[65,66]. Besides recognizing histone acetylation, BRD4 also identifies and binds to the acetylated lysine residues of non-histone. Shi et al.[67,68] discovered that BRD4 identifies Tip60-diacetylated of Twist and thereby constructing an activated Twist/BRD4/P-TEFb/RNA-Pol II complex at the WNT5A promoter and enhancer in breast cancer. BRD4 also functions as an atypical kinase to directly phosphorylate Serine 2 of the CTD of RNA POL II, implicating BRD4 as a regulator of transcription[69]. Recently, BRD4 also phosphorylates c-MYC at Thr58, resulting in MYC ubiquitination and degradation, suggesting BRD4 negatively regulates MYC level[70]. Overall, BRD4 possesses a pivotal role in the regulation of transcription and protein stabilization.
BRD4 plays an oncogenic role and is a potential target of therapy in various cancers. In CRPC,
Currently, there is a lack of BETis, including JQ1 approved by the FDA for clinic application due to dose-limiting toxicity. Given that combination treatment is a classic strategy to reduce the monotherapy dosage, Mao et al.[80] proposed that the PLK1 inhibitor GSK461364A could synergistically combine with BRD4 inhibitor JQ1 in the treatment of CRPC. The co-inhibition of BRD4 and PLK1 resulted in delayed cell growth, substantial cell apoptosis, and catastrophic cell cycle arrest in aggressive human CRPC cells. The significant improvement of efficacy in combining a PLK1 inhibitor and BRD4 inhibitor suggests a novel therapy for clinical trials.
Epigenetic erasers
Though epigenetic markers in post-translational modifications on histones are covalently linked to DNA, they are not permanently bound to the structure. Epigenetic erasers are a group of enzymes that maintain the ability to oppose the activity of writers and catalyze the removal of epigenetic alternations. This removal relieves its effect on transcription, resulting in the modulation of gene expression[17]. In the section, we emphasize the enzyme responsible for removing methyl and acetyl groups while discussing its role in the prostate and introducing therapeutic tactics.
HDAC
In contrast to histone acetyltransferase transferring acetyl group to histones, histone deacetylases (HDACs) remove acetyl groups from histones, resulting in a more condensed form of chromatin and gene silencing. To date, four HDAC classes have been identified in humans[81-83]. Class I HDACs, consisting of HDACs 1, 2, 3, and 8, are mainly localized in the nucleus and expressed in most tissues. Class II, consists of HDACs 4, 5, 6, 7, 9, and 10, are localized both in the nucleus and the cytoplasm. Class III HDACs are homologs of yeast silent information regulator 2 and consist of SIRT 1-7. Class IV HDAC consists of HDAC 11. Class I, II, and IV HDACs have a zinc coordinated active site, whereas Class III HDACs are dependent on coenzyme nicotinamide adenine dinucleotide for deacetylase activity.
HDAC role in PCa and therapeutic approaches
In cancer cells, high expression of HDACs results in the deacetylation of histone proteins, which causes DNA to be wrapped tightly by histones, thereby inhibiting gene expression. If the affected genes are tumor suppressors, the neoplastic proliferation of cells and cancer may result[83]. It has been reported that Class I HDACs (HDAC 1, 2, and 3) are highly expressed in PCa, specifically in CRPC[84,85]. Additionally, evidence has shown HDACs play a positive role in regulating the AR protein level and its transcriptional activity[86-88]. Therefore, it seems that HDACs could exhibit opposing pro- and anti-tumorigenic roles in PCa cells. In addition, HDAC inhibition could induce cell cycle arrest, apoptosis, autophagy, and reactive oxygen species generation[82,89]. The support from these discoveries has led to the initiation of several clinical trials of HDACs inhibitors in PCa treatment, including vorinostat, pracinostat, panobinostat, and romidepsin. However, none were recommended to continue phase III trials due to either toxicity or disease progression[82]. In summary, the function of HDAC in PCa and whether HDAC could be an effective target in the treatment of PCa is still ambiguous and requires further investigation to reach a conclusion.
Demethylase of histones
Histone lysine demethylases (KDMs) are a class of enzymes that can remove methyl groups from nucleic acids, proteins, and specifically histones. The first human KDM was reported in 2004[90].To date, several lysine-specific demethylase isoforms were discovered and characterized. Since their discovery, KDMs have been found to be deregulated in various cancers, such as non-small cell lung, breast, colorectal, pancreatic, etc.[87,91]. In PCa, KDMs may act as either tumor suppressors or oncogenes, which is dependent on the genes regulated by the KDMs.
Recently, Gao et al.[92] found that KDM1A is demethylation of FOXA1 at K270, and methylation of this residue decreases FOXA1 stability and activity. Inhibition of KDM1A, therefore, induces FOXA1 instability and results in FOXA1 chromatin dissociation, thus leading to loss of AR transcriptional activity. Consistent with this finding are several previously completed studies[93-95], which have demonstrated that KDM1A is required for the AR transcriptional activity regulation, both AR and AR variants, confirming its involvement in the progression of PCa. Interestingly, a recent study[96] showed that KDM1A could promote the survival of PCa cells independently of its demethylase function. This effect is explained by the activation of a lethal PCa gene network in collaboration with KDM1A’s binding protein, ZNF217. Numerous KDM1A inhibitors, such as TCP, ORY-1001, GSK-2879552, IMG-7289, INCB059872, CC-90011, and ORY-2001, have been reported and are presently being investigated in clinical trials for cancer treatment [Table 1][97]. Several have exhibited significantly improved potency and selectivity. In addition to KDM1A, KDM3A[94], KDM4A/4B[98-101], and KDM6A/6B[102] were also identified as coactivators of AR and play critical roles in PCa progression, thus characterizing them as potential therapeutic targets. These findings highlight the roles of KDMs in PCa initiation and progression, suggesting that targeting KDMs’ activity may provide a new strategy for PCa treatment.
Chromatin remodelers
Due to the budding advancements in high-throughput epigenomic approaches, visualizing chromatin structures and how their alternations result in disease development and progression has become an increased area of study[103]. Chromatin remodeling can be defined as the rearrangement of chromatin from a condensed state to a transcriptionally accessible state[104]. This rearrangement allows for transcription factors or DNA binding proteins to access DNA and control gene expression. This section will focus on chromatin remodeling as a compelling target for PCa therapeutic approaches.
ASF1A
Anti-silencing function 1A histone chaperone (ASF1A) is a major isoform of ASF1, a small histone chaperone of the H3/H4 family and conserved from yeast and human cells[105]. As the major isoform of ASF1 in human cells, ASF1A is ubiquitously expressed in all tissues and throughout the cell cycle[106]. The elevated expression of ASF1A positively correlates with the level of H3K56Ac[107], which is a mark of newly replicated chromatin as well as replication-independent histone replacement. ASF1A contributes to the resistance of DNA damage tolerance because of its ability to promote double-strand break (DSB) repair by non-homologous end joining. ASF1A deficiency and loss will render cells more sensitive to DSBs. For example, knockout ASF1A leads to the introduction of DSBs, which sensitizes cancer cells to radiotherapy, chemotherapy, and immunotherapy[108,109]. ASF1A has emerged as an oncogenic driver. Regarding several cancer cases, ASF1A accumulation is a general characteristic that occurs in tumorigenesis[110]. ASF1A is highly expressed in prostate cancer cells, and its overexpression is associated with poor prognosis in cancer patients[104,111-113]. Some reports have shown that blocking the expression of ASF1A by RNA interference[108,111], small inhibitors, and chemotherapy drugs[110] can effectively inhibit the proliferation and growth of tumors and improve the sensitivity to anti-cancer drugs and immunotherapy[108,109].
CAF-1
Histone chaperone chromatin assembly factor-1 (CAF-1) is composed of p150 large unit (CHAF1A), p60 middle unit (CHAF1B), and p48 small unit (RbAp48) and is involved in the deposition of (H3-H4)2 tetramer onto DNA[114]. During replication, CAF-1 receives (H3-H4)2 tetramer from another histone chaperone ASF1A and then deposits the histone onto the newly synthesized daughter DNA strands[114,115]. It has been reported that the dysregulation of histone assembly is closely associated with certain human diseases such as cancer[116]. Indeed, CAF-1 has been shown to be a marker of proliferating cells[117], and depletion of CAF-1 induces cell death, possibly due to the activation of DNA damage response pathway[118]. Specifically, in PCa, the middle unit of CAF-1 is a prognostic marker of adverse outcomes for patients[111], and inhibition of ASF1A suppresses the growth of PCa[119]. These interesting results raise the possibility that targeting chromatin assembly in PCa is a potential treatment for PCa patients.
SAFB1
SAFB1 (scaffold attachment factor B1) is a nonenzymatic architectural component of the chromatin that was first identified to bind adenine- and thymine-rich scaffold/matrix attachment (S/MAR) regions[120] to divide the genome into 5-200 kb topological domains. SAFB1 was previously assumed to mediate chromatin looping to modulate long-range chromatin interactions and higher-order chromatin structure[119]. SAFB1 is a component of the DNA damage response and cooperates with histone acetylation to allow for efficient gH2AX spreading and genotoxic stress signaling. SAFB1 undergoes a highly dynamic exchange at damaged chromatin in a poly (ADP-ribose)-polymerase 1- and poly (ADP-ribose)-dependent manner and is required for unperturbed cell cycle checkpoint activation and guarding cells against replicative stress[121]. Meanwhile, SAFB1 regulates RNA polymerase II-dependent transcription of targeted genes[119]. There is a potent transcriptional repression domain at the C-terminal region of SAFB1, which mediates the transcriptional repression activity. Particularly, SAFB1 binds to nuclear receptors[122,123] and suppresses immune regulators and apoptotic genes[124]. SAFB1 attenuates ERα transcriptional activity via its interaction with the ERα DNA-binding domain in a ligand-independent manner[125]. Low levels of SAFB1 were found to correlate with worse outcomes in breast cancer patients[126]. In addition, SAFB1 is also reduced with disease progression in a cohort of human PCa, including metastatic tumors. SAFB1 binds to AR and is phosphorylated by the MST1 (Hippo homolog) serine-threonine kinase, an AR repressor, and MST1 localization to AR-dependent promoters is inhibited by depletion of SAFB1. Meanwhile, SAFB1 interacts with Enhancer of Zeste 2 Polycomb Repressive Complex 2 (EZH2) at ARE of chromatin. Knockdown of SAFB1 in androgen-dependent LNCaP cells results in upregulation of AR and PSA levels, stimulating the growth of cultured cells and subcutaneous xenografts and promoting a more aggressive phenotype, which is consistent with a negative AR regulatory function[127]. Collectively, SAFB1 functions as a tumor suppressor in both breast cancer and PCa.
Epigenetics and genetic instability
Epigenetics and genetics have been described as separate entities, participating in carcinogenesis via independent mechanisms[128]. However, recent publications have unveiled crosstalk that occurs between genome and epigenome factors that could produce novel therapeutic strategies in PCa[127].
Microsatellites are highly polymorphic, short-tandem repeat sequences dispersed throughout the genome[128]. The instability of these repeats at multiple loci can result in mismatch repair errors and other genetic issues. Loss of heterozygosity (LOH) has been reported to strongly correlate with increasing malignancy in prostate carcinoma[129]. Recently it has been documented that chromosomal instability, including MSI/LOH, has been categorized as a distinct type of genetic instability characteristic in regards to prostate cancer[130]. Epigenetic processes such as hypermethylation of tumor suppressors, histone modification, and hypomethylation of oncogenes have been documented to eventually create genetic instability in the forms of MSI, LOH, allelic loss, single nucleotide polymorphisms (SNPs), and chromosomal aberrations. DNA methylation alterations could induce loss of heterozygosity and lead to a progression in prostate cancer[129]. It has been reported ten-eleven translocase 2 (TET2), enzyme-mediated DNA demethylation, exhibits high mutation rates (10%-20%) and extensive loss of heterozygosity (~60%) in metastatic prostate tumors. Genome-wide association studies have also shown increased PCa risk linked to an intergenic TET2-proximal SNP (rs7679673)[131]. Additionally, Baylin and Jones have reported that cancers with hypermethylated MGMT are susceptible to genetic mutations in critical genes such as p53 or KRAS. MLH1, a mismatch repair gene, plays an important role in genomic instability. It has been reported that promoter hypermethylation results in loss of function of this gene and causes MSI in several cancers[132].
Regarding histone modification, deregulated Polycomb Repressor Complex 2 mediated epigenetic modifications have been shown to cause genetic instability, malignancy, and cancer development through abnormal tumor suppressor gene expression, DNA damage response, and DNA replication[133]. BRD4 acetylates histone H3 at the K122 residue, and this thereby perturbs a salt bridge, leading to nucleosome instability[134]. It has been recently documented that targeting genetic instability with possible PARP could be utilized as a novel therapeutic approach in prostate cancer treatment. Epigenetic changes, such as DNA hyper- and hypomethylation, can cause genetic instability, such as LOH/MSI, in various cancer types. Multiple genetic and epigenetic abnormalities in PCa suggest that co-targeting both epigenetic changes and genetic instability could become a novel therapeutic strategy in PCa treatment.
CONCLUSION
AR has been a critical target for the treatment of PCa, and while ADT has been effective in preventing cancer cell proliferation, progression to a more aggressive phenotype is inevitable. In this review, we discussed the various epigenetic changes which contribute to the further advancement and progression of PCa via the activation of various oncogenic pathways. We also explored novel therapeutic approaches established by our lab and drug treatment strategies that have demonstrated impactful success. Due to recent discoveries in the understanding of the mechanisms of maintained AR signaling in castration-resistance PCa, targeting these epigenetic changes that facilitate AR target gene activation has a highly possible and promising potential in developing novel therapeutic approaches. Though whether targeting these factors’ stability will produce toxic or ineffective effects is obscure, pre-clinical trial data gathered and documented by our lab indicates that clinical trial participation could result in highly efficient and optional treatment methods.
DECLARATIONS
Authors’ contributionsMade substantial contributions to conception, material articulation, and editing of the review: Jones K
Provided administrative, technical, and material support: Zhang Y
Provided material support: Kong Y, Wang R, Farah E, Li C, Wang X, Zhang Z, Wang J, Mao F
Contributed to the conception and design of the review: Liu X, Liu J
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work was supported by NIH grants R01 CA157429 (Liu X), R01 CA192894 (Liu X), R01 CA196835
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
2. Juárez Soto A, Caballero Cobos R, Campanario Pérez R, et al. [Abiraterone in castration resistant prostate cancer]. Arch Esp Urol 2018;71:651-63.
3. Kong Y, Zhang Y, Mao F, et al. Inhibition of EZH2 Enhances the antitumor efficacy of metformin in prostate cancer. Mol Cancer Ther 2020;19:2490-501.
4. Zhang Z, Cheng L, Li J, et al. Inhibition of the Wnt/β-Catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res 2018;78:3147-62.
5. Chen X, Liu J, Cheng L, et al. Inhibition of noncanonical Wnt pathway overcomes enzalutamide resistance in castration-resistant prostate cancer. Prostate 2020;80:256-66.
6. Kong Y, Cheng L, Mao F, et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J Biol Chem 2018;293:14328-41.
7. Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028-38.
8. Wang R, Sun Y, Li L, et al. Preclinical study using malat1 small interfering RNA or androgen receptor splicing variant 7 degradation enhancer ASC-J9® to suppress enzalutamide-resistant prostate cancer progression. Eur Urol 2017;72:835-44.
9. Yamamoto Y, Loriot Y, Beraldi E, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin Cancer Res 2015;21:1675-87.
10. Farah E, Li C, Cheng L, et al. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J Biol Chem 2019;294:8543-54.
11. Biswas S, Rao CM. Epigenetic tools (the writers, the readers and the erasers) and their implications in cancer therapy. Eur J Pharmacol 2018;837:8-24.
12. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol 2010;28:1057-68.
13. Hake SB, Xiao A, Allis CD. Linking the epigenetic “language” of covalent histone modifications to cancer. Br J Cancer 2004;90:761-9.
14. Tuorto F, Herbst F, Alerasool N, et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J 2015;34:2350-62.
15. Jurkowska RZ, Anspach N, Urbanke C, et al. Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res 2008;36:6656-63.
16. Ashour N, Angulo JC, Andrés G, et al. A DNA hypermethylation profile reveals new potential biomarkers for prostate cancer diagnosis and prognosis. Prostate 2014;74:1171-82.
17. Angulo JC, Andrés G, Ashour N, Sánchez-Chapado M, López JI, Ropero S. Development of castration resistant prostate cancer can be predicted by a DNA hypermethylation profile. J Urol 2016;195:619-26.
18. Moritz R, Ellinger J, Nuhn P, et al. DNA hypermethylation as a predictor of PSA recurrence in patients with low- and intermediate-grade prostate cancer. Anticancer Res 2013;33:5249-54.
19. Barry KH, Mohanty K, Erickson PA, et al. MYC DNA methylation in prostate tumor tissue is associated with gleason score. Genes (Basel) 2020;12:12.
20. Rauluseviciute I, Drabløs F, Rye MB. DNA hypermethylation associated with upregulated gene expression in prostate cancer demonstrates the diversity of epigenetic regulation. BMC Med Genomics 2020;13:6.
21. Mohammadi M, Irani S, Salahshourifar I, Hosseini J, Moradi A, Pouresmaeili F. The effect of hormone therapy on the expression of prostate cancer and multi-epigenetic marker genes in a population of iranian patients. Cancer Manag Res 2020;12:3691-7.
22. Gravina GL, Marampon F, Piccolella M, et al. Hormonal therapy promotes hormone-resistant phenotype by increasing DNMT activity and expression in prostate cancer models. Endocrinology 2011;152:4550-61.
23. Chen X, Skutt-Kakaria K, Davison J, et al. G9a/GLP-dependent histone H3K9me2 patterning during human hematopoietic stem cell lineage commitment. Genes Dev 2012;26:2499-511.
24. Poulard C, Bittencourt D, Wu DY, Hu Y, Gerke DS, Stallcup MR. A post-translational modification switch controls coactivator function of histone methyltransferases G9a and GLP. EMBO Rep 2017;18:1442-59.
25. Lee DY, Northrop JP, Kuo MH, Stallcup MR. Histone H3 lysine 9 methyltransferase G9a is a transcriptional coactivator for nuclear receptors. J Biol Chem 2006;281:8476-85.
26. Chin HG, Estève PO, Pradhan M, et al. Automethylation of G9a and its implication in wider substrate specificity and HP1 binding. Nucleic Acids Res 2007;35:7313-23.
27. Rathert P, Dhayalan A, Murakami M, et al. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol 2008;4:344-6.
28. Chen MW, Hua KT, Kao HJ, et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res 2010;70:7830-40.
29. Hua KT, Wang MY, Chen MW, et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol Cancer 2014;13:189.
30. Lee JS, Kim Y, Kim IS, et al. Negative regulation of hypoxic responses via induced Reptin methylation. Mol Cell 2010;39:71-85.
31. Lee JS, Kim Y, Bhin J, et al. Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc Natl Acad Sci U S A 2011;108:13510-5.
32. Lee SH, Kim J, Kim WH, Lee YM. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene 2009;28:184-94.
33. Casciello F, Al-Ejeh F, Miranda M, et al. G9a-mediated repression of CDH10 in hypoxia enhances breast tumour cell motility and associates with poor survival outcome. Theranostics 2020;10:4515-29.
34. Kang J, Shin SH, Yoon H, et al. FIH is an oxygen sensor in ovarian cancer for G9a/GLP-driven epigenetic regulation of metastasis-related genes. Cancer Res 2018;78:1184-99.
35. Ding J, Li T, Wang X, et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab 2013;18:896-907.
36. Dong C, Yuan T, Wu Y, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013;23:316-31.
37. Wang YF, Zhang J, Su Y, et al. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat Commun 2017;8:274.
38. Dutta A, Le Magnen C, Mitrofanova A, Ouyang X, Califano A, Abate-Shen C. Identification of an NKX3.1-G9a-UTY transcriptional regulatory network that controls prostate differentiation. Science 2016;352:1576-80.
39. Segovia C, San José-Enériz E, Munera-Maravilla E, et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat Med 2019;25:1073-81.
40. Guler GD, Tindell CA, Pitti R, et al. Repression of stress-induced LINE-1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer Cell 2017;32:221-237.e13.
41. Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 2002;298:1039-43.
42. Yu J, Yu J, Mani RS, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010;17:443-54.
43. Kim E, Kim M, Woo DH, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013;23:839-52.
44. Sanulli S, Justin N, Teissandier A, et al. Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol Cell 2015;57:769-83.
45. He A, Shen X, Ma Q, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev 2012;26:37-42.
46. Shi B, Liang J, Yang X, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol 2007;27:5105-19.
47. Lee ST, Li Z, Wu Z, et al. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol Cell 2011;43:798-810.
48. Gonzalez ME, Moore HM, Li X, et al. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc Natl Acad Sci U S A 2014;111:3098-103.
49. Xu K, Wu ZJ, Groner AC, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012;338:1465-9.
50. Kim J, Lee Y, Lu X, et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep 2018;25:2808-20.e4.
51. Rodriguez-Vida A, Galazi M, Rudman S, Chowdhury S, Sternberg CN. Enzalutamide for the treatment of metastatic castration-resistant prostate cancer. Drug Des Devel Ther 2015;9:3325-39.
52. Bai Y, Zhang Z, Cheng L, et al. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide resistance in castration-resistant prostate cancer. J Biol Chem 2019;294:9911-23.
53. Welti J, Sharp A, Brooks N, et al. SU2C/PCF International Prostate Cancer Dream Team. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov 2021;11:1118-37.
54. Xia C, Tao Y, Li M, Che T, Qu J. Protein acetylation and deacetylation: an important regulatory modification in gene transcription (review). Exp Ther Med 2020;20:2923-40.
55. Zhong J, Ding L, Bohrer LR, et al. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer Res 2014;74:1870-80.
56. Fu M, Wang C, Reutens AT, et al. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J Biol Chem 2000;275:20853-60.
57. Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017;550:128-32.
58. Jin L, Garcia J, Chan E, et al. Therapeutic Targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res 2017;77:5564-75.
59. Liu J, He D, Cheng L, et al. p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment in prostate cancer. Oncogene 2020;39:3939-51.
60. Ghosh AK, Steele R, Ray RB. Knockdown of MBP-1 in human prostate cancer cells delays cell cycle progression. J Biol Chem 2006;281:23652-7.
61. Haynes SR, Dollard C, Winston F, Beck S, Trowsdale J, Dawid IB. The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res 1992;20:2603.
63. Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999;399:491-6.
64. Jiang YW, Veschambre P, Erdjument-Bromage H, et al. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc Natl Acad Sci U S A 1998;95:8538-43.
65. Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 2005;19:523-34.
66. Yang Z, Yik JH, Chen R, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 2005;19:535-45.
67. Shi J, Wang Y, Zeng L, et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014;25:210-25.
68. Shi J, Cao J, Zhou BP. Twist-BRD4 complex: potential drug target for basal-like breast cancer. Curr Pharm Des 2015;21:1256-61.
69. Devaiah BN, Lewis BA, Cherman N, et al. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci U S A 2012;109:6927-32.
70. Devaiah BN, Mu J, Akman B, et al. MYC protein stability is negatively regulated by BRD4. Proc Natl Acad Sci U S A 2020;117:13457-67.
71. Pawar A, Gollavilli PN, Wang S, Asangani IA. Resistance to BET inhibitor leads to alternative therapeutic vulnerabilities in castration-resistant prostate cancer. Cell Rep 2018;22:2236-45.
72. Nagarajan S, Hossan T, Alawi M, et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep 2014;8:460-9.
73. Faivre EJ, McDaniel KF, Albert DH, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020;578:306-10.
74. Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature 2010;468:1067-73.
75. Shu S, Polyak K. BET bromodomain proteins as cancer therapeutic targets. Cold Spring Harb Symp Quant Biol 2016;81:123-9.
76. Gilan O, Rioja I, Knezevic K, et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020;368:387-94.
77. Dai X, Gan W, Li X, et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 2017;23:1063-71.
78. Zhou B, Hu J, Xu F, et al. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J Med Chem 2018;61:462-81.
79. Raina K, Lu J, Qian Y, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2016;113:7124-9.
80. Mao F, Li J, Luo Q, et al. Plk1 inhibition enhances the efficacy of BET epigenetic reader blockade in castration-resistant prostate cancer. Mol Cancer Ther 2018;17:1554-65.
81. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 2014;6:a018713.
82. Rana Z, Diermeier S, Hanif M, Rosengren RJ. Understanding failure and improving treatment using HDAC inhibitors for prostate cancer. Biomedicines 2020;8:22.
83. Kaushik D, Vashistha V, Isharwal S, Sediqe SA, Lin MF. Histone deacetylase inhibitors in castration-resistant prostate cancer: molecular mechanism of action and recent clinical trials. Ther Adv Urol 2015;7:388-95.
84. Weichert W, Röske A, Gekeler V, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer 2008;98:604-10.
85. Graça I, Pereira-Silva E, Henrique R, Packham G, Crabb SJ, Jerónimo C. Epigenetic modulators as therapeutic targets in prostate cancer. Clin Epigenetics 2016;8:98.
86. Gao L, Alumkal J. Epigenetic regulation of androgen receptor signaling in prostate cancer. Epigenetics 2010;5:100-4.
87. Welsbie DS, Xu J, Chen Y, et al. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res 2009;69:958-66.
88. Sato S, Katsushima K, Shinjo K, et al. Histone deacetylase inhibition in prostate cancer triggers miR-320-mediated suppression of the androgen receptor. Cancer Res 2016;76:4192-204.
89. Robey RW, Chakraborty AR, Basseville A, et al. Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol Pharm 2011;8:2021-31.
90. Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004;119:941-53.
91. Crea F, Sun L, Mai A, et al. The emerging role of histone lysine demethylases in prostate cancer. Mol Cancer 2012;11:52.
92. Gao S, Chen S, Han D, et al. Chromatin binding of FOXA1 is promoted by LSD1-mediated demethylation in prostate cancer. Nat Genet 2020;52:1011-7.
93. Regufe da Mota S, Bailey S, Strivens RA, et al. LSD1 inhibition attenuates androgen receptor V7 splice variant activation in castration resistant prostate cancer models. Cancer Cell Int 2018;18:71.
94. Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005;437:436-9.
95. Wissmann M, Yin N, Müller JM, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 2007;9:347-53.
96. Sehrawat A, Gao L, Wang Y, et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc Natl Acad Sci U S A 2018;115:E4179-88.
97. Fang Y, Liao G, Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol Oncol 2019;12:129.
98. Wilson S, Fan L, Sahgal N, Qi J, Filipp FV. The histone demethylase KDM3A regulates the transcriptional program of the androgen receptor in prostate cancer cells. Oncotarget 2017;8:30328-43.
99. Kim TD, Jin F, Shin S, et al. Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J Clin Invest 2016;126:706-20.
100. Chu CH, Wang LY, Hsu KC, et al. KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor. J Med Chem 2014;57:5975-85.
101. Duan L, Rai G, Roggero C, et al. KDM4/JMJD2 histone demethylase inhibitors block prostate tumor growth by suppressing the expression of AR and BMYB-regulated genes. Chem Biol 2015;22:1185-96.
102. Stief SM, Hanneforth AL, Weser S, et al. Loss of KDM6A confers drug resistance in acute myeloid leukemia. Leukemia 2020;34:50-62.
103. Gao Y, Gan H, Lou Z, Zhang Z. Asf1a resolves bivalent chromatin domains for the induction of lineage-specific genes during mouse embryonic stem cell differentiation. Proc Natl Acad Sci U S A 2018;115:E6162-71.
104. Abascal F, Corpet A, Gurard-Levin ZA, et al. Subfunctionalization via adaptive evolution influenced by genomic context: the case of histone chaperones ASF1a and ASF1b. Mol Biol Evol 2013;30:1853-66.
105. Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009;459:113-7.
106. Li F, Huang Q, Luster TA, et al. In vivo epigenetic CRISPR screen identifies Asf1a as an immunotherapeutic target in Kras-mutant lung adenocarcinoma. Cancer Discov 2020;10:270-87.
107. Lee KY, Im JS, Shibata E, Dutta A. ASF1a promotes non-homologous end joining repair by facilitating phosphorylation of MDC1 by ATM at double-strand breaks. Mol Cell 2017;68:61-75.e5.
108. Wang C, Chang JF, Yan H, et al. A conserved RAD6-MDM2 ubiquitin ligase machinery targets histone chaperone ASF1A in tumorigenesis. Oncotarget 2015;6:29599-613.
109. Henrique R. ASF1A in gastric and colorectal cancer: on the hinge between genetics and epigenetics? EBioMedicine 2017;21:45-6.
110. Liang X, Yuan X, Yu J, et al. Histone chaperone asf1a predicts poor outcomes for patients with gastrointestinal cancer and drives cancer progression by stimulating transcription of β-catenin target genes. EBioMedicine 2017;21:104-16.
111. Wu Y, Li X, Yu J, Björkholm M, Xu D. ASF1a inhibition induces p53-dependent growth arrest and senescence of cancer cells. Cell Death Dis 2019;10:76.
112. Im JS, Keaton M, Lee KY, Kumar P, Park J, Dutta A. ATR checkpoint kinase and CRL1βTRCP collaborate to degrade ASF1a and thus repress genes overlapping with clusters of stalled replication forks. Genes Dev 2014;28:875-87.
113. Sauer PV, Gu Y, Liu WH, et al. Mechanistic insights into histone deposition and nucleosome assembly by the chromatin assembly factor-1. Nucleic Acids Res 2018;46:9907-17.
114. Buschbeck M, Hake SB. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol 2017;18:299-314.
115. Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol 2013;20:14-22.
116. Polo SE, Theocharis SE, Klijanienko J, et al. Chromatin assembly factor-1, a marker of clinical value to distinguish quiescent from proliferating cells. Cancer Res 2004;64:2371-81.
117. Nabatiyan A, Krude T. Silencing of chromatin assembly factor 1 in human cells leads to cell death and loss of chromatin assembly during DNA synthesis. Mol Cell Biol 2004;24:2853-62.
118. Staibano S, Mascolo M, Mancini FP, et al. Overexpression of chromatin assembly factor-1 (CAF-1) p60 is predictive of adverse behaviour of prostatic cancer. Histopathology 2009;54:580-9.
119. Garee JP, Oesterreich S. SAFB1's multiple functions in biological control-lots still to be done! J Cell Biochem 2010;109:312-9.
120. Renz A, Fackelmayer FO. Purification and molecular cloning of the scaffold attachment factor B (SAF-B), a novel human nuclear protein that specifically binds to S/MAR-DNA. Nucleic Acids Res 1996;24:843-9.
121. Altmeyer M, Toledo L, Gudjonsson T, et al. The chromatin scaffold protein SAFB1 renders chromatin permissive for DNA damage signaling. Mol Cell 2013;52:206-20.
122. Debril MB, Dubuquoy L, Feige JN, et al. Scaffold attachment factor B1 directly interacts with nuclear receptors in living cells and represses transcriptional activity. J Mol Endocrinol 2005;35:503-17.
123. Oesterreich S, Zhang Q, Hopp T, et al. Tamoxifen-bound estrogen receptor (ER) strongly interacts with the nuclear matrix protein HET/SAF-B, a novel inhibitor of ER-mediated transactivation. Mol Endocrinol 2000;14:369-81.
124. Hammerich-Hille S, Kaipparettu BA, Tsimelzon A, et al. SAFB1 mediates repression of immune regulators and apoptotic genes in breast cancer cells. J Biol Chem 2010;285:3608-16.
125. Hammerich-Hille S, Bardout VJ, Hilsenbeck SG, Osborne CK, Oesterreich S. Low SAFB levels are associated with worse outcome in breast cancer patients. Breast Cancer Res Treat 2010;121:503-9.
126. Mukhopadhyay NK, Kim J, You S, et al. Scaffold attachment factor B1 regulates the androgen receptor in concert with the growth inhibitory kinase MST1 and the methyltransferase EZH2. Oncogene 2014;33:3235-45.
127. Sugiura M, Sato H, Kanesaka M, et al. Epigenetic modifications in prostate cancer. Int J Urol 2021;28:140-9.
128. Suzuki H, Komiya A, Aida S, et al. Microsatellite instability and other molecular abnormalities in human prostate cancer. Jpn J Cancer Res 1995;86:956-61.
129. Hügel A, Wernert N. Loss of heterozygosity (LOH), malignancy grade and clonality in microdissected prostate cancer. Br J Cancer 1999;79:551-7.
130. Koochekpour S. Genetic and epigenetic changes in human prostate cancer. Iran Red Crescent Med J 2011;13:80-98.
131. Kamdar S, Isserlin R, Van der Kwast T, et al. Exploring targets of TET2-mediated methylation reprogramming as potential discriminators of prostate cancer progression. Clin Epigenetics 2019;11:54.
132. Li K, Luo H, Huang L, Luo H, Zhu X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int 2020;20:16.
133. Veneti Z, Gkouskou KK, Eliopoulos AG. Polycomb repressor complex 2 in genomic instability and cancer. Int J Mol Sci 2017;18:1657.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Jones K, Zhang Y, Kong Y, Farah E, Wang R, Li C, Wang X, Zhang ZZ, Wang J, Mao F, Liu X, Liu J. Epigenetics in prostate cancer treatment. J Transl Genet Genom 2021;5:341-56. http://dx.doi.org/10.20517/jtgg.2021.19
AMA Style
Jones K, Zhang Y, Kong Y, Farah E, Wang R, Li C, Wang X, Zhang ZZ, Wang J, Mao F, Liu X, Liu J. Epigenetics in prostate cancer treatment. Journal of Translational Genetics and Genomics. 2021; 5(3): 341-56. http://dx.doi.org/10.20517/jtgg.2021.19
Chicago/Turabian Style
Jones, Katelyn, Yanquan Zhang, Yifan Kong, Elia Farah, Ruixin Wang, Chaohao Li, Xinyi Wang, ZhuangZhuang Zhang, Jianlin Wang, Fengyi Mao, Xiaoqi Liu, Jinghui Liu. 2021. "Epigenetics in prostate cancer treatment" Journal of Translational Genetics and Genomics. 5, no.3: 341-56. http://dx.doi.org/10.20517/jtgg.2021.19
ACS Style
Jones, K.; Zhang Y.; Kong Y.; Farah E.; Wang R.; Li C.; Wang X.; Zhang ZZ.; Wang J.; Mao F.; Liu X.; Liu J. Epigenetics in prostate cancer treatment. J. Transl. Genet. Genom. 2021, 5, 341-56. http://dx.doi.org/10.20517/jtgg.2021.19
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 19 clicks
Cite This Article 19 clicks

 Like This Article 2
likes
Like This Article 2
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.