
Developmental and epileptic encephalopathies after negative or inconclusive genetic testing: what is next?

 , ...
, ... Abstract
The redefinition of classical electroclinical syndromes and the emergence of neurogenetics has led to a revolution in the field of developmental and epileptic encephalopathies (DEEs). In this context, advances in genetic techniques are leading to the final diagnosis of a large proportion of patients with DEE. However, up to 50% of patients with DEE remain undiagnosed. For patients with uncertain genetic etiology, there is a pressing need for the implementation of new targeted treatments and precision medicine. In some undiagnosed patients, genetic reanalysis with further in-depth or reverse phenotyping are valuable diagnostic tools to clarify new variants of uncertain significance. In other cases, the implementation of new bioinformatic algorithms is required for the update and reassessment of previously generated genetic data. Moreover, many other clinical tools have been developed for the management of patients of DEEs after a negative or inconclusive genetic testing. In this review, we highlight advances and limitations of new diagnostic strategies used in DEE patients without a known genetic etiology. Finally, we provide a wide perspective on aspects that will need further research, especially in non-Mendelian inheritance DEEs, such as those related to somatic mosaicism of the central nervous system or epigenetic and oligogenic mechanisms.
Keywords
INTRODUCTION
The field of epilepsy genetics has emerged in clinical practice and is rapidly evolving in the last years. In this setting, the main group of epilepsies where a genetic etiology can be found are the developmental and epileptic encephalopathies (DEEs), defined as wide electroclinical syndromes characterized by epilepsy, developmental delay or regression or intellectual disability, an abnormal EEG, and other possible neurological or systemic manifestations[1]. Next-generation sequencing (NGS) provided a rapid increment of gene discoveries in human disorders. The actual prevalence of DEEs is not known yet, although it has been estimated that the overall annual incidence of single-gene epilepsies is around 1 per 2100 live births[2]. This recent epidemiological research on genetic epilepsies led to elucidating some of the most common genes associated with DEEs, such as SCN1A, KCNQ2, PCDH19, CDKL5, SCN2A, and SCN8A, among others[3]. Currently, genetic testing is mandatory for determining precision medicine treatments and to avoid unnecessary and potentially harmful diagnostic examinations and medications. A genetic diagnosis might also provide genetic counseling and relevant information on the natural history of a condition, as well as the access to new clinical trials or patient support groups[4].
Multiple lines of evidence showing poor genotype-phenotype correlations in classical electroclinical syndromes have triggered a redefinition of the traditional concept “epileptic encephalopathy”, as well as a revolution in the field of DEEs over the last few years[5]. At the clinical level, these changes promoted the application of new procedures when studying genetic and phenotypic heterogeneity, e.g., by expanding the clinical spectrum related to each gene in a process known as reverse phenotyping[6]. Importantly, since a large proportion of genes related to epilepsy are also associated with autism or intellectual disability, among others[7], the transition towards the conceptual framework “DEEs” has made it possible to redefine epilepsy as an additional symptom of neurodevelopmental diseases. Advances in genetic diagnostic techniques have also led to the confirmed diagnosis of a large proportion of patients with DEEs. However, at least a relevant proportion of patients - estimated as 50% - remain undiagnosed[8]. In most cases, patients remain undiagnosed due to insufficient genetic testing. In others, variants of uncertain significance have been identified after further in-depth phenotyping. The update of genetic studies and/or the reanalysis of genetic data with modern computational tools has proved essential as our knowledge of the disease prognosis and the availably of targeted treatments increases. However, there are still many aspects related to the course of clinical diagnostic of DEEs that need to evolve to reach precision treatments, especially in non-Mendelian inherited neurodevelopmental disorders, such as those related to mitochondrial genome, somatic mosaicism of the central nervous system, or epigenetic alterations[9,10].
The aims of this narrative review are: (1) to understand the limitations in the diagnosis of classical epileptic syndromes and their reframing in light of the most recent advances in genetic epidemiology; (2) to describe the reverse phenotyping strategy and its importance in the clinical practice regarding patients with DEEs; (3) to discuss the management of patients with DEE and negative or inconclusive genetic testing; and (4) to understand the limitations of the current diagnostic approach for these patients and future perspectives.
CONCEPTUAL FRAMEWORK SHIFT OF “EPILEPTIC ENCEPHALOPATHY”
Developmental and epileptic encephalopathies are a heterogeneous and complex group of diseases that fall under the general umbrella of neurodevelopmental disorders, in which epilepsy and cognitive/behavioral problems are combined in varying proportions, in some cases associated with other comorbidities. There is a group of “classical” epileptic encephalopathies, such as West, Lennox-Gastaut, or Dravet syndromes, which were defined as characteristic electro-clinical syndromes before the etiology was fully understood. However, these syndromes are currently undergoing a revisited definition[5].
The concept of “epileptic encephalopathy”, which the International League Against Epilepsy defines as the process whereby “epileptic activity and seizures generate cognitive and/or behavioral problems beyond those expected for the etiology”, has become obsolete[11]. Substantial clinical data suggest that neurodevelopmental impairment in classical epileptic encephalopathies start before epilepsy onset and progress after epilepsy remission. In light of these data, current views of DEEs consider that epileptic activity in classical epileptic encephalopathies plays only a partial or limited role in the neurodevelopment of the patient. In those cases, it is established that epilepsy is more an epiphenomenon of the etiological process (in most cases, genetic), and that it is the abnormal neurodevelopmental trajectory which plays the leading role in the pathophysiological process. Thus, the concept of “developmental and epileptic encephalopathies” has emerged, in which it is accepted that neurodevelopmental problems are partly due to the etiology and partly due to the epileptic process[12].
Most classically defined electroclinical syndromes have a poor genotype-phenotype correlation, with polygenic risk and multiple genes producing the same syndrome (genetic heterogeneity) as well as with pleiotropic risk alleles - multiple syndromes produced by the same gene (phenotypic heterogeneity). Thus, syndromes such as West or Lennox-Gastaut are now considered common pathways of expression of multiple etiologies, so more accurate phenotyping of these patients, beyond the classical triads, will be essential to reach a diagnosis[13].
However, there are other electroclinical syndromes that could be considered successful examples in terms of genotype-phenotype correlation, such as Dravet syndrome. It is caused by pathogenic variants in SCN1A in about 85%-90% of cases, showing a haploinsufficiency mechanism. In addition, the diagnosis of Dravet syndrome has been refined in recent years, as many genes traditionally included in the syndrome, such as PCDH19 or KCNA2, now have their own specific syndrome, with differential characteristics from classical Dravet[14,15]. Thus, the redefinition of the concept of epileptic encephalopathies, together with a poorly genotype-phenotype correlation in the classical electroclinical syndromes, triggered this paradigm shift in the study of DEEs.
DIFFICULTIES IN GENOTYPE-PHENOTYPE CORRELATION
Phenotypic expansion
As a result of the increased number of patients analyzed with platforms that include a high number of genes, the phenotypic spectrum related to specific genetic conditions has increasingly expanded. Taking the SCN1A gene as example, some years ago variants located in this locus were associated with two distinct and unique phenotypes: Dravet syndrome and genetic epilepsy with febrile seizures plus (GEFS+). GEFS+ is a dominantly inherited epilepsy syndrome in which individuals show a predisposition to spontaneous seizures or seizures triggered by fever and other factors, such as emotional excitement, without refractory epilepsy or neurodevelopmental disturbances. In contrast, in Dravet syndrome, initial febrile seizures progress to severe refractory epilepsy after the second or third year, showing neurodevelopmental problems[16].
Currently, clinical findings on SCN1A variants suggest that, in addition to the full spectrum of phenotypes between GEFS+ and Dravet syndrome, there are also substantially different SCN1A-associated epilepsy phenotypes that do not fall within the GEFS+/Dravet spectrum[17]. Moreover, SCN1A variants may be associated with other neurological phenotypes that do not include epilepsy, such as familial hemiplegic migraine[18]. This scenario, characterized by a wide variability in epilepsy phenotypes as well as non-epileptic neurological phenotypes, has emerged for most epilepsy-associated genes[19]. It is likely that some, but not all, of the phenotypic variability is explained by gene variant-specific functional effects. For example, SCN1A variants associated with hemiplegic migraine typically show gain of function. Other genetic and environmental modifiers are likely to account for much of the remaining phenotypic variability, but these factors remain difficult to be fully established and characterized. This has been shown in different research works with functional studies, with relevant translational implications in genetic-phenotypic correlations, such as the case of some biallelic variants in SCN1B[20].
Identification of new genetic variants
Targeted analyses focused on the identification of epilepsy-related genes can be separated into two types: (1) those that compare huge cohorts of people with epilepsy with healthy individuals and search for a higher proportion of common genetic variants among people with epilepsy, i.e., genome-wide association studies (GWAS)[21]; and (2) those that look for rare deleterious variants, which might give an explanation of patient’s clinical presentation in order to be considered as causative[22]. Milder phenotypes are more convenient for GWAS analysis, while severe phenotypes (DEEs) are suitable for the second strategy. De novo pathogenic variants, which in most cases produce reproductive difficulties and are thus very rarely found in the general healthy population, are often the most robust candidate variants[23].
GWAS studies do not try to search for disease causes. This approach normally tries to elucidate the common genetic variants that confer a higher predisposition to epilepsy. This screening approach has been valuable in understanding the gene-related architecture of epilepsy[24]. Among these studies, scarce variants have obtained genome-wide significance, and those variants explain a tiny percentage of the general phenotypic variation[25]. Thus, these research strategies show low clinical relevance. Future progress in this direction will need larger numbers and less heterogeneity. In recent years, “polygenic risk scoring” strategies are being developed attempting to determine the genetic load of common variants that together generate more frequent and milder phenotypes, such as adult focal epilepsies or idiopathic generalized epilepsies[26].
Studies using next generation sequencing techniques to identify rare deleterious variants in deeply phenotyped patients with severe neurodevelopmental disorders with epilepsy provide more useful opportunities for new epilepsy-related genes discoveries[27]. These approaches analyzed the epilepsy-related disorders from two different perspectives. The first perspective involves the analysis of wide samples of patients with broad phenotypes, with or without associated epilepsy, exemplified by the Deciphering Developmental Disorders (DDD) study[28]. The second involves the analysis of samples of individuals who concretely presented epilepsy, such as the Epi4K consortium study[29]. It is important to distinguish between these two approaches, since, in the first one, epilepsy represents one of the symptoms included in a global neurodevelopmental disorder, while, in the second one, epilepsy is the main selection criterion and additional symptoms are presented as co-morbidities.
The illustrative case of STXBP1
Deleterious de novo variants in STXBP1 were firstly published in four patients with early-onset epilepsy, with Ohtahara-syndrome features[30]. Therefore, STXBP1 was firstly labeled as an “Ohtahara syndrome gene”. Subsequent publications revealed that STXBP1 variants were associated with many other epilepsy syndromes, such as Dravet, West or Lennox-Gastaut syndrome[31]. Currently, STXBP1 phenotypic spectrum is considered even broader, as a “neurodevelopmental disease-related gene”, since 11 patients of the DDD study (out of 4293 families reported) had de novo deleterious STXBP1 variants[32]. From this cohort, three patients had no history of seizures and two others presented epilepsy in late childhood[33]. Altogether, the evidence suggests that there are few epilepsy-specific genes, and most of them are generally associated with global neurodevelopmental diseases, causing both intellectual disability and autism spectrum disorders[34].
REVERSE PHENOTYPING
Reverse phenotyping refers to the refinement of phenotypes based on the interpretation of genetic data. The strategy was firstly utilized in sarcoidosis patients and their phenotyping, and it is a promising new approach in many other heterogeneous diseases, such as epilepsy or neurodevelopmental disorders[35] [Figure 1].
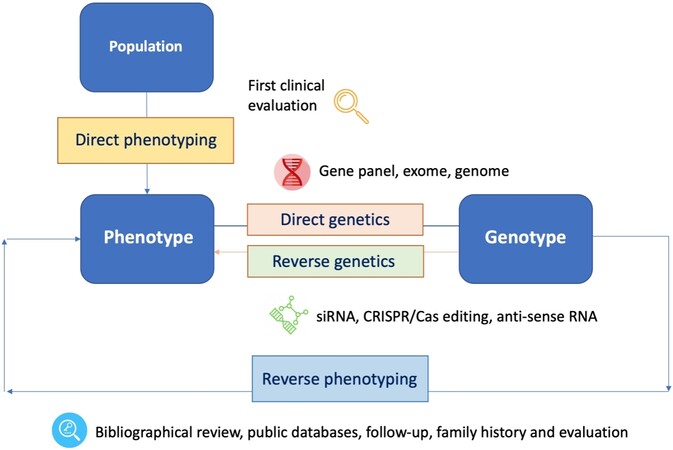
Figure 1. Diagram depicting the concepts of “direct genetics”, i.e., the “genotype to phenotype” approach; “reverse genetics”, i.e., the analysis of the impact of an induced variation in a specific gene for a gene function with different techniques (CRISPR/Cas, etc.); and “reverse phenotyping”, i.e., the clinical assessment driven by genetic results. siRNA: small interfering RNA.
The picture of PCDH19-related epilepsy
The PCDH19 gene was initially included in the group of genes related to Dravet syndrome as it was associated with early-onset febrile and prolonged seizures before two years of life and is usually associated with cognitive and behavioral disturbances[36]. Patients with pathogenic variants could be included into a wide Dravet or “Dravet-like” phenotype. However, over time these patients were found to have very specific characteristics. Most of the patients are girls, present seizures that occur in clusters, and have focal semiology. In addition, cognitive and behavioral disorders are slightly different (including autistic spectrum features without motor abnormalities), and the drugs that show efficacy are also different. Through a reverse phenotyping process (from the gene to the syndrome and not from the syndrome to the gene, as routinely performed), it was observed that there was a specific PCDH19-related epilepsy syndrome that could be distinguished from the classical Dravet syndrome. Currently, it is known that PCDH19 is among the 10 most frequent causes of genetic epilepsy with onset in the first two years of life causing a very distinctive syndrome. This translates into different treatments[37]. For example, levetiracetam is a drug with low efficacy in Dravet syndrome and very high efficacy in PCDH19-related epilepsy[38].
Platforms to work with for reverse phenotyping
A major goal in the field of clinical genetics is to better understand the conundrums of genotype-phenotype correlations. A detailed review of the recent literature and publicly available databases is necessary when dealing with variants of uncertain significance[39]. This process has been facilitated in recent years by the emergence of bioinformatic tools, such as the R package VarformPDB, which organize genes and variants associated to a condition, phenotype, or clinical feature from public databases such as OMIM (Online Mendelian Inheritance in Man), Orphanet, ClinVar, Human Phenotype Ontology (HPO), UniProt (Universal Protein Resource), and abstracts from PubMed[40]. Moreover, the creation of huge biobanks associating electronic medical record (EMR) data and genetics has led to improved clinical characterization of variant carriers. In addition to deep clinical phenotyping, multi-omics strategies can be employed through simultaneous compilation of biomarkers and samples from patients[40]. Multiple innovative projects have been made to ease EMR-based genomic research, such as the creation of the Precision Medicine Initiative Cohort Programme, which allow both “direct genetic” and “reverse phenotyping” implementations. The use of EHRs for genomic research presents multiple advantages, as large samples can be recruited without the previous time-consuming and economic burden[40,41].
The clinical genetics community employs numerous tools to address reverse phenotyping, including DECIPHER, an open-access database that hosts a collection of tests for more than 37,000 patients with genetic conditions to compare genotype-phenotype data[42]. Additional examples, such as the Matchmaker Exchange or GeneMatcher, serve as database mining using the association of genotype and phenotype profiles[43]. On the other hand, VarSome is a bioinformatic platform with tools aiming to compile information needed for variant analysis[44]. All these platforms allow the neurologist and geneticist to perform direct and inverse phenotyping of patients, facilitating the interpretation of variants and the diagnosis and management of patients.
HOW TO MANAGE PATIENTS WITH “NEGATIVE” GENETIC TESTING?
Fast and correct diagnosis is essential for appropriate patient management. However, patients and families with DEEs usually spend more than five or six years in the so-called diagnostic odyssey. This diagnostic journey often implies several evaluations to specialists and diagnostic tests (sometimes invasive), and it can entail relevant social and individual disturbances. Unfortunately, even with the broad number of diagnostic techniques available, at least 50% of DEE patients do not receive a genetic diagnosis[45]. Although less than 3% of genetic diseases currently have an FDA-approved therapy, rigorous genetic diagnosis is crucial for clinical care in many aspects (e.g., genetic counseling, prognosis and family planning, and choosing specific traditional treatments[46]) and for patient and family quality of life (end of diagnostic odyssey, emotional peace, and access to patient advocacy and supporting groups)[47].
Most of the worldwide population does not have access to sufficient genetic testing, which represents the most challenging problem for affected individuals with DEEs. Several countries have recognized that patients with DEEs face systematic difficulties to access medical attention and diagnostic techniques[48,49]. Some of these problems derive from the infrequent prevalence, which increases complexity in diagnosis and appropriate management. These include low awareness, no guidelines for diagnosis, and minimal medical knowledge in DEEs among physicians. There are additional system barriers, such as limited access to genetic testing in some populations. In countries such as Spain, studies show that access to genetic testing in patients with epilepsy remains limited, especially among adults with DEEs[50].
Challenges in the analysis and interpretation of variants of uncertain significance
Even after our rapid increment in knowledge about the diverse genetic mechanisms, we do not understand yet most of the genomic variations. The American College of Medical Genetics and Genomics (ACMG) defines a variant of uncertain significance (VUS) as a genetic variant with a probability of pathogenicity between 10% and 90%[51]. The proportion of VUS recognized in every report is variable, as are some factors, such as the indication, the size of the genetic region, and the current knowledge of a function in a particular protein[52]. This increment in VUS produces difficulties in patient outcomes and is a major challenge in the correct diagnosis of DEEs.
The clinical validation of genetic testing outcomes is complex to establish. In several examples, there are phenotypic expansions of already known disease-gene correlations. When numbers analyzed increase, the detection of atypical phenotypic presentations of DEE grow, expanding the phenotype and the clinical variability associated with that specific gene. Usually, the severe forms of the disease are better established, while milder forms will remain hidden until genetic studies begin to be requested in a larger sample of patients with less severe epilepsy forms[53].
Our knowledge about the gene variant-disease relationship is continuously growing; approximately 300 new identified associations or new genes are reported each year[54]. These newly described associations may result from genes that had never been related to a genetic condition (new disease gene identification) or that were previously related to a different disease (new phenotypic association). Some laboratories report variants in genes of uncertain significance (GUS), requiring the search for other individuals with variants in the that GUS and similar or overlapping clinical features to aid this candidate gene as a disease-causing gene and ultimately a diagnosis[55]. Some studies elucidate that around 6000-13,000 Mendelian diseases remain to be defined[55]. Although this suggests a huge potential for the identification of new disease-gene associations, those sometimes cannot be discover using the current diagnostic strategy, thus requiring in some cases the implementation of functional studies to elucidate the gene variant-disease association[56]. In light of these findings, the evaluation and interpretation of studies in DEE patients requires continuous reevaluation and analysis. These are outlined schematically in Figure 2. Additionally, recent disease-focused specifications for ACMG guidelines might be helpful in the interpretation of some clinical scenarios, regarding variants (especially VUS) with specific genes/disorders[57].
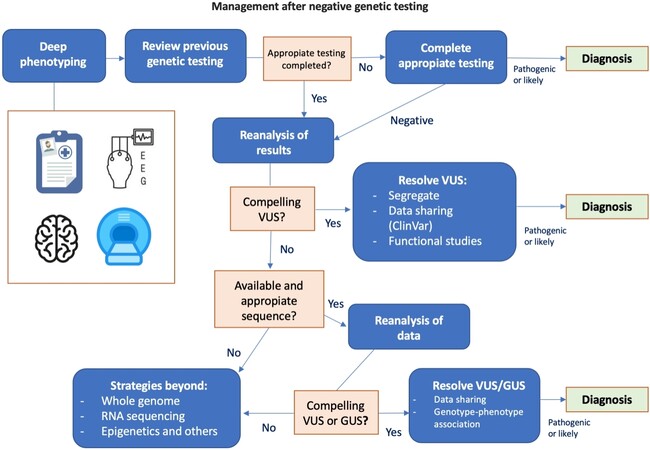
Figure 2. Schematic of the diagnostic procedure in developmental and epileptic encephalopathie patients presenting negative genetic testing. The first step is phenotyping and clinical reevaluation of the patient with electroclinical data, neuroimaging, phenotyping of relatives, or other tests such as cognitive and behavioral profiling. This leads to a diversion point when the procedure will include completing the study and/or reanalyzing previous studies, assessing variants of uncertain significance (VUS), reevaluating the data with different bioinformatics algorithms, or analyzing genes of uncertain significance (GUS). Sometimes the limitations of sequencing make it necessary to employ other techniques, such as whole genome, transcriptomic (RNA) sequencing, or methylation analysis techniques, among others.
Suitability and sufficiency of previous genetic testing
“Negative genetic testing” in patients with DEE is often the result of scarce workup. Thus, a large proportion of negative genetic outcomes in this population results from insufficient screening or obsolete evaluation requiring an update[58]. There are three types of genetic studies that are currently considered insufficient in all types of DEEs: karyotyping, single gene sequencing, and array-CGH. Karyotyping is rarely indicated in epilepsy (only in cases of suspected ring chromosomes, such as 20 or 14) and should not be considered as the unique study to be performed in any case[59]. Regarding single- or two-gene sequencing, there are also few clinical scenarios where it can be efficient. The most characteristic are Dravet syndrome or tuberous sclerosis, entities where the predictive capacity for a specific genetic diagnosis (SCN1A or TSC1/2) is high[60]. However, in most cases, if this first approach is negative, large panels or exome sequencing will be appropriate. Array-CGH is a test that has been widely used in DEE patients, but its diagnostic yield is of only about 10%[61]. Therefore, its negative study should never be considered as sufficient in these patients. Whole exome sequencing (WES) is currently considered the most appropriate technique for genetic diagnosis in DEE, with a diagnostic yield of up to 50%[62], which improves if trio strategies are used (WES also performed in the parents to be able to segregate identified variants of interest), with bioinformatics algorithms for the detection of copy number variations (CNV, which would also replace array-CGH in a proportion of cases) or the detection of nucleotide expansions[63]. Moreover, some studies have shown that resequencing strategies in previously specified pediatric epilepsy cohorts could significantly increase the diagnostic yield, through the analysis of exon-intron boundaries and selected non-coding and deep intronic variants, as well as single nucleotide variants and small INDEL (up to 220 bps) and CNVs using NGS[64].
Reanalysis of previous negative sequencing data
Since the current paradigm is not designed for DEE patients without diagnoses, there is a need to broaden our approach to clinical care for these patients. An integral part of this evaluation is the recurrent reanalysis of sequencing data and data sharing to facilitate the probability of reaching a correct genetic diagnosis. In-depth phenotyping of individuals with an undiagnosed DEE, sometimes including the family, is critical, and it is most useful when using an international and systematized nomenclature, such as HPO[65]. Of note, this diagnostic strategy includes atypical phenotypes, clinical expansions, combination of more than one disease, and genetic associations of newly discovered diseases[66].
Firstly, the patient phenotype is deeply analyzed. Subsequently, the strategy of previous tests should be reviewed, in order to look for pitfalls that did not lead to a diagnosis. Previous tests should be reviewed to reveal whether a broad approach (in genes and conditions) was properly performed. For patients with convincing VUS, we would start by searching for new publications with information showing pathogenicity (e.g., in ClinVar or HGMD) or benign variation (e.g., high frequency in gnomAD). Some additional information might come from segregation or functional studies[67].
Nowadays, it is not only important to carry out quality genetic studies, but also to have the possibility to reanalyze them after a period (maximum every 3-5 years). Exome sequencing that is negative today may be positive in two years. The clinician should demand access to sequences and regular reanalysis of patient data. In many clinical fields, WES is available as a funded test for most of the patients with complex phenotypes[66]. However, it is only 30%-50% successful[67]. Although it is not established which percentage of individuals carry pathogenic variants in the protein-coding part of the genome (exome), it is likely to be much higher than this diagnostic yield. In fact, a negative exome is considered a point of start for the patient without a diagnosis, as it has been documented that there is a high probability that reanalysis will identify diagnoses both in old and new genes[68].
Reanalysis of exome data provide additional diagnostics for some reasons[55,61,68,69]:
(1) Immediate reanalysis of exome data with geneticist-clinician collaboration has been reported to reach a diagnosis in known disease genes in 10% of undiagnosed patients. This gap in genetic diagnostic finds its origin in a poor clinical interpretation of genetic data when interaction between geneticists and clinical neurologists is lacking.
(2) In a similar way in which new information on disease-associated variants can assist in the reevaluation of VUS identified in known genes, the approximately 300 disease-gene associations newly described every year may identify variants that were not evaluated when the genetic testing was performed.
(3) Clinicians may elucidate these new disease-gene correlations on their own. It has been demonstrated that discovery of new genes through recurrent analysis of exome data can double the diagnostic yield[69].
(4) Reanalysis collaborating with the laboratory using distinct bioinformatics approaches can reach a diagnosis which was missed using the previous strategies.
(5) Reanalysis allows the clinician to modulate different thresholds (e.g., quality or rarity) and use different bioinformatic tools. It depends on some clinical scenarios, such as mosaic variants mimicking autosomal recessive pattern of inheritance.
(6) RNA sequencing methods can play a role in some specific cases, with the identification of pathogenic variants that remain unnoticed or doubtful after WES[70].
LIMITATIONS AND FUTURE PERSPECTIVES
Currently, there is a broad spectrum of genetic diagnostic tools suitable for clinical assessment of DEEs. However, all have technical limitations. Clinicians must embrace that, even if the best selected genetic test for their patient had been performed, a genetic alteration cannot be certainly ruled out; they may only reduce its probability. For instance, although NGS (next-generation sequencing) techniques have improved our DNA sequencing ability, there are still difficulties with GC-rich regions, homology, or pseudogenes. Additionally, although microarrays (array-CGH) show greatly improved ability to confirm CNVs, their limited resolution on pre-defined sequences means that some diagnoses can still be missed[71]. Moreover, there are technical limitations of NGS that are typical in the bioinformatics algorithms used to align genomic data to a reference sequence, and annotation and filtering strategies may affect which variants are subsequently identified for analysis[72]. Whole genome or trio genome (including parents) sequencing (WGS) studies are starting to be used, with some publications showing promising results in DEEs without diagnosis[73]. However, the major difficulty associated with WGS is the arduous mass of information provided, which must be properly mined and interpreted through close geneticist-clinical collaboration. The roles of many genes and complex genome variations are still undetermined. Thus, although WGS can produce a large volume of data, most of them may be misleading or useless at the moment. Then, the genetic testing strategy selected for a patient with an undiagnosed DEEs should be analyzed in detail. This will help us to better understand with which probability are current methodologies in genetic screening missing disease mechanisms[74].
Millions of individuals with rare diseases have undergone targeted genetic assessment, and more than 100,000 patients worldwide have undergone an NGS sequencing technique (especially whole exome sequencing approach) during their clinical care[75]. However, access to most of these data is difficult, if not impossible, because healthcare is a highly regulated environment due to policies on personal data privacy and potential solutions are difficult and costly. Even in research, taking in account that individuals usually agree to share their whole data, access is usually a very limited process to ensure compliance. Strategies must be developed to capitalize on previous as well as future data and fully exploit the powerful collective data in order to assist the interpretation of genetic variation for patient care[76]. The balance between data sharing and maintaining confidentiality will ultimately impact our diagnostic capacity in the future.
Finally, we must not lose sight that current clinical-grade NGS and associated bioinformatics analyses cannot confirm genetic variation associated with non-Mendelian inheritance, such as tissue (brain)-specific somatic mosaicism (very frequent in epilepsy and already widely linked to cortical developmental malformations), epigenetic abnormalities, oligogenic mechanisms, or gene-environment interactions[77]. For instance, pathogenic variant mosaicism might not be easily identified depending on the accuracy of the selected approach, and detection is especially hampered for tissue-specific mosaicism (especially in the central nervous system) and low-level somatic mutations. Epigenetic alterations, such as methylation defects, uniparental disomy, and altered expression profiles of the parents of origin at a locus, require specific testing approaches. In addition, there are currently no clinical-implemented strategies to detect oligogenic or complex inheritance (even with polygenic risk scores), where the cumulative effect of different common and rare variants, together with environmental triggers in the context of a complex disease, results in the clinical picture of a specific patient[78]. The study of DEEs caused by these mechanisms is currently limited and will warrant attention and research in the next future.
DECLARATIONS
Authors’ contributionsMade substantial contributions to conception, material articulation, and editing of the review: Aledo-Serrano A, Sánchez-Alcudia R, Tolerano R, Garcia-Morales I, Beltrán-Corbellini A, del Pino I, Gil-Nagel A
Provided administrative, technical, and material support: Aledo-Serrano A, Sánchez-Alcudia R
Provided material support: Sánchez-Alcudia R, Tolerano R, Garcia-Morales I, Beltrán-Corbellini A,
Contributed to the conception and design of the review: Aledo-Serrano A, Sanchez-Alcudia R, Gil-Nagel A
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Ellis CA, Petrovski S, Berkovic SF. Epilepsy genetics: clinical impacts and biological insights. Lancet Neurol 2020;19:93-100.
2. Symonds JD, Zuberi SM, Stewart K, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain 2019;142:2303-18.
3. Symonds JD, McTague A. Epilepsy and developmental disorders: next generation sequencing in the clinic. Eur J Paediatr Neurol 2020;24:15-23.
4. Hebbar M, Mefford HC. Recent advances in epilepsy genomics and genetic testing. F1000Res 2020;9:185.
5. Kalser J, Cross JH. The epileptic encephalopathy jungle - from Dr West to the concepts of aetiology-related and developmental encephalopathies. Curr Opin Neurol 2018;31:216-22.
6. Johnson MR, Shorvon SD. Heredity in epilepsy: neurodevelopment, comorbidity, and the neurological trait. Epilepsy Behav 2011;22:421-7.
7. Shankar R, Perera B, Thomas RH. Epilepsy, an orphan disorder within the neurodevelopmental family. J Neurol Neurosurg Psychiatry 2020;91:1245-7.
8. Striano P, Minassian BA. From genetic testing to precision medicine in epilepsy. Neurotherapeutics 2020;17:609-15.
9. Heinzen EL. Somatic variants in epilepsy - advancing gene discovery and disease mechanisms. Curr Opin Genet Dev 2020;65:1-7.
10. Jesus-Ribeiro J, Pires LM, Melo JD, et al. Genomic and epigenetic advances in focal cortical dysplasia types I and II: a scoping review. Front Neurosci 2020;14:580357.
11. Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010;51:676-85.
12. Korff CM, Brunklaus A, Zuberi SM. Epileptic activity is a surrogate for an underlying etiology and stopping the activity has a limited impact on developmental outcome. Epilepsia 2015;56:1477-81.
13. Specchio N, Curatolo P. Developmental and epileptic encephalopathies: what we do and do not know. Brain 2021;144:32-43.
14. Steel D, Symonds JD, Zuberi SM, Brunklaus A. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia 2017;58:1807-16.
15. Aledo-Serrano A, Mingorance A. [Analysis of the family impact and needs of Dravet's syndrome in Spain]. Rev Neurol 2020;70:75-83.
16. Brunklaus A, Zuberi SM. Dravet syndrome - from epileptic encephalopathy to channelopathy. Epilepsia 2014;55:979-84.
17. Riva A, Coppola A, Balagura G, et al. Temporal-parietal-occipital epilepsy in GEFS+ associated with SCN1A mutation. Epileptic Disord 2021;23:397-401.
18. Scheffer IE, Nabbout R. SCN1A-related phenotypes: epilepsy and beyond. Epilepsia 2019;60 Suppl 3:S17-24.
19. Guerrini R, Balestrini S, Wirrell EC, Walker MC. Monogenic epilepsies: disease mechanisms, clinical phenotypes, and targeted therapies. Neurology 2021;10.1212/WNL.0000000000012744.
20. Scala M, Efthymiou S, Sultan T, et al. Homozygous SCN1B variants causing early infantile epileptic encephalopathy 52 affect voltage-gated sodium channel function. Epilepsia 2021;62:e82-7.
21. Song M, Liu J, Yang Y, Lv L, Li W, Luo XJ. Genome-wide meta-analysis identifies two novel risk loci for epilepsy. Front Neurosci 2021;15:722592.
22. Symonds JD, Zuberi SM. Genetics update: monogenetics, polygene disorders and the quest for modifying genes. Neuropharmacology 2018;132:3-19.
23. Perucca P, Bahlo M, Berkovic SF. The genetics of epilepsy. Annu Rev Genomics Hum Genet 2020;21:205-30.
24. Takata A, Nakashima M, Saitsu H, et al. Comprehensive analysis of coding variants highlights genetic complexity in developmental and epileptic encephalopathy. Nat Commun 2019;10:2506.
25. Greenberg DA, Stewart WC. How should we be searching for genes for common epilepsy? Epilepsia 2012;53 Suppl 4:72-80.
26. Leu C, Stevelink R, Smith AW, et al. Epi25 Consortium. Polygenic burden in focal and generalized epilepsies. Brain 2019;142:3473-81.
27. Liu X, Shen Q, Zheng G, et al. Gene and phenotype expansion of unexplained early infantile epileptic encephalopathy. Front Neurol 2021;12:633637.
28. Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017;542:433-8.
29. Allen AS, Berkovic SF, Cossette P, et al. Epi4K Consortium, Epilepsy Phenome/Genome Project. De novo mutations in epileptic encephalopathies. Nature 2013;501:217-21.
30. Milh M, Villeneuve N, Chouchane M, et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011;52:1828-34.
31. Wild B, Nelson S. STXBP1-related developmental and epileptic encephalopathy. Pediatr Neurol Briefs 2019;33:6.
32. Abramov D, Guiberson NGL, Burré J. STXBP1 encephalopathies: clinical spectrum, disease mechanisms, and therapeutic strategies. J Neurochem 2021;157:165-78.
33. Suri M, Evers JMG, Laskowski RA, et al. DDD Study. Protein structure and phenotypic analysis of pathogenic and population missense variants in STXBP1. Mol Genet Genomic Med 2017;5:495-507.
34. Liu X, Sun X, Sun C, et al. Prevalence of epilepsy in autism spectrum disorders: a systematic review and meta-analysis. Autism 2021:13623613211045029.
35. Swietlik EM, Prapa M, Martin JM, et al. 'There and back again'-forward genetics and reverse phenotyping in pulmonary arterial hypertension. Genes (Basel) 2020;11:1408.
36. Samanta D. PCDH19-related epilepsy syndrome: a comprehensive clinical review. Pediatr Neurol 2020;105:3-9.
37. Aledo-Serrano Á, Del Ser T, Gil-Nagel A. Antiseizure medication withdrawal in seizure-free patients with PCDH19-related epilepsy: a multinational cohort survey. Seizure 2020;80:259-61.
38. Sadleir LG, Kolc KL, King C, et al. Levetiracetam efficacy in PCDH19 girls clustering epilepsy. Eur J Paediatr Neurol 2020;24:142-7.
39. Chakravorty S, Hegde M. Gene and variant annotation for mendelian disorders in the era of advanced sequencing technologies. Annu Rev Genomics Hum Genet 2017;18:229-56.
40. Sayers EW, Beck J, Bolton EE, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 2021;49:D10-7.
41. Byrne S, Enright N, Delanty N. Precision therapy in the genetic epilepsies of childhood. Dev Med Child Neurol 2021; doi: 10.1111/dmcn.14929.
42. Sobreira NLM, Arachchi H, Buske OJ, et al. Matchmaker Exchange Consortium. Matchmaker exchange. Curr Protoc Hum Genet 2017;95:9.31.1-9.31.15.
43. Wohler E, Martin R, Griffith S, et al. PhenoDB, GeneMatcher and VariantMatcher, tools for analysis and sharing of sequence data. Orphanet J Rare Dis 2021;16:365.
44. Azzariti DR, Hamosh A. Genomic data sharing for novel mendelian disease gene discovery: the matchmaker exchange. Annu Rev Genomics Hum Genet 2020;21:305-26.
45. Møller RS, Hammer TB, Rubboli G, Lemke JR, Johannesen KM. From next-generation sequencing to targeted treatment of non-acquired epilepsies. Expert Rev Mol Diagn 2019;19:217-28.
46. Aledo-Serrano Á, Gómez-Iglesias P, Toledano R, et al. Sodium channel blockers for the treatment of epilepsy in CDKL5 deficiency disorder: findings from a multicenter cohort. Epilepsy Behav 2021;118:107946.
47. Wu AC, McMahon P, Lu C. Ending the diagnostic odyssey - is whole-genome sequencing the answer? JAMA Pediatr 2020;174:821-2.
48. Espinosa-Jovel C, Toledano R, Aledo-Serrano Á, García-Morales I, Gil-Nagel A. Epidemiological profile of epilepsy in low income populations. Seizure 2018;56:67-72.
49. Raspa M, Moultrie R, Toth D, Haque SN. Barriers and facilitators to genetic service delivery models: scoping review. Interact J Med Res 2021;10:e23523.
50. Aledo-Serrano A, García-Morales I, Toledano R, et al. Diagnostic gap in genetic epilepsies: a matter of age. Epilepsy Behav 2020;111:107266.
51. Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.
52. Frebourg T. The challenge for the next generation of medical geneticists. Hum Mutat 2014;35:909-11.
53. Achkar CM, Harrer M, Smith L, et al; GABRB2 Working Group. Characterization of the GABRB2-associated neurodevelopmental disorders. Ann Neurol 2021;89:573-86.
54. Bamshad MJ, Nickerson DA, Chong JX. Mendelian gene discovery: fast and furious with no end in sight. Am J Hum Genet 2019;105:448-55.
55. Strande NT, Riggs ER, Buchanan AH, et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am J Hum Genet 2017;100:895-906.
56. Carvill GL, Helbig KL, Myers CT, et al. Damaging de novo missense variants in EEF1A2 lead to a developmental and degenerative epileptic-dyskinetic encephalopathy. Hum Mutat 2020;41:1263-79.
57. Harrison SM, Biesecker LG, Rehm HL. Overview of specifications to the ACMG/AMP variant interpretation guidelines. Curr Protoc Hum Genet 2019;103:e93.
58. Moyses-Oliveira M, Yadav R, Erdin S, Talkowski ME. New gene discoveries highlight functional convergence in autism and related neurodevelopmental disorders. Curr Opin Genet Dev 2020;65:195-206.
59. Gago-Veiga AB, Toledano R, García-Morales I, Pérez-Jiménez MA, Bernar J, Gil-Nagel A. Specificity of electroclinical features in the diagnosis of ring chromosome 20. Epilepsy Behav 2018;80:215-20.
60. Peron A, Au KS, Northrup H. Genetics, genomics, and genotype-phenotype correlations of TSC: insights for clinical practice. Am J Med Genet C Semin Med Genet 2018;178:281-90.
61. Scala M, Bianchi A, Bisulli F, et al. Advances in genetic testing and optimization of clinical management in children and adults with epilepsy. Expert Rev Neurother 2020;20:251-69.
62. Jiang T, Gao J, Jiang L, et al. Application of trio-whole exome sequencing in genetic diagnosis and therapy in chinese children with epilepsy. Front Mol Neurosci 2021;14:699574.
63. Bayat A, Bayat M, Rubboli G, Møller RS. Epilepsy syndromes in the first year of life and usefulness of genetic testing for precision therapy. Genes (Basel) 2021;12:1051.
64. Amadori E, Scala M, Cereda GS, et al. Targeted re-sequencing for early diagnosis of genetic causes of childhood epilepsy: the Italian experience from the 'beyond epilepsy' project. Ital J Pediatr 2020;46:92.
65. Leblond CS, Le TL, Malesys S, et al. Operative list of genes associated with autism and neurodevelopmental disorders based on database review. Mol Cell Neurosci 2021;113:103623.
66. Zacher P, Mayer T, Brandhoff F, et al. The genetic landscape of intellectual disability and epilepsy in adults and the elderly: a systematic genetic work-up of 150 individuals. Genet Med 2021;23:1492-7.
67. Zou D, Wang L, Liao J, et al. Genome sequencing of 320 Chinese children with epilepsy: a clinical and molecular study. Brain 2021:awab233.
68. Myers KA, Johnstone DL, Dyment DA. Epilepsy genetics: current knowledge, applications, and future directions. Clin Genet 2019;95:95-111.
69. Eldomery MK, Coban-Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med 2017;9:26.
70. Ghosh SG, Scala M, Beetz C, et al. Undiagnosed Diseases Network. A relatively common homozygous TRAPPC4 splicing variant is associated with an early-infantile neurodegenerative syndrome. Eur J Hum Genet 2021;29:271-9.
71. Basel-Salmon L, Orenstein N, Markus-Bustani K, et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet Med 2019;21:1443-51.
72. Zahrani H, Siriwardena K, Young D, Lehman A, Horvath GA, Goez H. Genomics in Cerebral Palsy phenotype across the lifespan: comparison of diagnostic yield between children and adult population. Mol Genet Metab ;2021:S1096-7192(21)00758.
73. Bozkurt T, Alanay Y, Isik U, Sezerman U. Re-analysis of whole-exome sequencing data reveals a novel splicing variant in the SLC2A1 in a patient with GLUT1 Deficiency Syndrome 1 accompanied by hemangioma: a case report. BMC Med Genomics 2021;14:197.
74. Dunn P, Albury CL, Maksemous N, et al. Next generation sequencing methods for diagnosis of epilepsy syndromes. Front Genet 2018;9:20.
75. Woerner AC, Gallagher RC, Vockley J, Adhikari AN. The use of whole genome and exome sequencing for newborn screening: challenges and opportunities for population health. Front Pediatr 2021;9:663752.
76. Vamathevan J, Birney E. A review of recent advances in translational bioinformatics: bridges from biology to medicine. Yearb Med Inform 2017;26:178-87.
77. Kobow K, Baulac S, von Deimling A, Lee JH. Molecular diagnostics in drug-resistant focal epilepsy define new disease entities. Brain Pathol 2021;31:e12963.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Aledo-Serrano A, Sánchez-Alcudia R, Toledano R, García-Morales I, Beltrán-Corbellini, del Pino I, Gil-Nagel A. Developmental and epileptic encephalopathies after negative or inconclusive genetic testing: what is next?. J Transl Genet Genom 2021;5:443-55. http://dx.doi.org/10.20517/jtgg.2021.40
AMA Style
Aledo-Serrano A, Sánchez-Alcudia R, Toledano R, García-Morales I, Beltrán-Corbellini, del Pino I, Gil-Nagel A. Developmental and epileptic encephalopathies after negative or inconclusive genetic testing: what is next?. Journal of Translational Genetics and Genomics. 2021; 5(4): 443-55. http://dx.doi.org/10.20517/jtgg.2021.40
Chicago/Turabian Style
Aledo-Serrano, Angel, Rocío Sánchez-Alcudia, Rafael Toledano, Irene García-Morales, Álvaro Beltrán-Corbellini, Isabel del Pino, Antonio Gil-Nagel. 2021. "Developmental and epileptic encephalopathies after negative or inconclusive genetic testing: what is next?" Journal of Translational Genetics and Genomics. 5, no.4: 443-55. http://dx.doi.org/10.20517/jtgg.2021.40
ACS Style
Aledo-Serrano, A.; Sánchez-Alcudia R.; Toledano R.; García-Morales I.; Beltrán-Corbellini.; del Pino I.; Gil-Nagel A. Developmental and epileptic encephalopathies after negative or inconclusive genetic testing: what is next?. J. Transl. Genet. Genom. 2021, 5, 443-55. http://dx.doi.org/10.20517/jtgg.2021.40
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 22 clicks
Cite This Article 22 clicks

 Like This Article 2
likes
Like This Article 2
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.