
Epilepsy and electroencephalography in Pitt-Hopkins syndrome


Abstract
Aim: Pitt-Hopkins syndrome (PTHS) is a rare neurodevelopmental disorder caused by mono-allelic loss of function variants of transcription factor 4 (TCF4), which plays a key role in early brain developmental and neuronal differentiation. Up to one-half of patients with PTHS will have epilepsy; however, little is known about the characteristic electroencephalogram (EEG) findings in this population. Because there is significant phenotypic overlap between PTHS and other neurodevelopmental disorders such as Angelman syndrome and Rett syndrome, which have characteristic EEG patterns, exploration of a potential EEG signature in patients with PTHS was warranted.
Methods: We conducted a retrospective review of clinical EEGs in patients with PTHS.
Results: In this cohort of patients with PTHS (n = 16), over half had abnormal EEGs; however, no characteristic EEG signature was identified. Further, all patients with epilepsy (5/16) had focal onset seizures with or without secondary generalization, and all five had focal abnormalities on EEG. There was no specific correlation between EEG results and developmental trajectories or age in our patient group, and there was no clear genotype-phenotype correlation.
Conclusion: Although a distinctive EEG signature was not identified, all individuals with epilepsy in our cohort had focal onset seizures with corresponding focal epileptiform discharges or focal slowing on EEG. Future studies are needed to fully elucidate the spectrum of EEG findings in PTHS and explore the pathogenesis of focal seizures in a disorder of neuronal differentiation and development.
Keywords
INTRODUCTION
Pitt-Hopkins syndrome (PTHS) is a haploinsufficiency syndrome caused by loss of function variants of transcription factor 4 (TCF4) on chromosome 18q21.2. It was first described in 1978 by Drs. Pitt and Hopkins, and TCF4 was identified as the causative gene in 2007[1]. PTHS is an ultra-orphan disease with an estimated prevalence of 1:225,000-1:300,000[2-4]. Clinically, patients with PTHS present with symptoms that are similar to other more common neurodevelopmental syndromes such as Angelman syndrome and Rett syndrome. Though PTHS patients often have characteristic facial features such as a narrow forehead, thin lateral eyebrows, wide nasal bridge, ridge, or tip, flared nasal alae, full cheeks, prominent midface, wide mouth, full lips, prominent cupid’s bow upper lip, and thickened or over-folded helices[1,2,5-7]. The developmental phenotype is relatively homogenous, with profound developmental disabilities including markedly delayed onset of ambulation and limited or absent speech[1,2]. Individuals with PTHS have impairments in adaptive behavior on standardized testing, with scores resembling profiles seen in patients with Angelman syndrome[8,9]. Patients with PTHS are also often affected by additional medical comorbidities, including early-onset myopia (54%-88%), chronic constipation (70%-83%), and unsteady gait with impaired mobility[1,3]. Brain imaging is often normal but may demonstrate non-specific findings such as corpus callosal dysplasia, bulging of the caudate heads, small hippocampi, temporal white matter hyperintensities, delayed myelination, hypoplasia of the cerebellum, or ventriculomegaly[1,2,10].
There are three types of paroxysmal events that PTHS patients may have: (1) abnormal breathing spells (e.g., hyperventilation with or without apnea); (2) motor stereotypies; and (3) epileptic seizures. Nearly half of patients with PTHS have spells of abnormal breathing, including hyperventilation, which may be followed by apnea with rapid onset of cyanosis[1]. Breathing spells are consistently only observed while awake and do not have an ictal electroencephalogram (EEG) correlate[11]. This suggests that breathing spells are not epileptic events but could be a behavioral phenomenon or related to autonomic dysfunction. Motor stereotypies are common and can include hand flapping, rocking, twisting, waving, or flicking hands, and repetitively handling objects[4]. The prevalence of epilepsy in PTHS is 37%-50%, with onset anytime between the first year of life and adulthood[4], but more commonly within the first decade of life (2). In PTHS patients with or without epilepsy, EEG can be normal or abnormal and can change over time[4]. Seizures can have variable semiology, including generalized tonic-clonic, atonic, focal motor, focal non-motor seizures, and rarely infantile spasms[1]. One case report identified a child with PTHS who had both migrating partial seizures of infancy and infantile spasms[12]. Apnea and hyperventilation spells are often misdiagnosed as seizures, and to add to the diagnostic challenge, patients may show apnea or over-breathing just prior to onset of a seizure[4].
A case series of 21 patients with PTHS and epilepsy found that the median age of seizure onset was two years and that focal and generalized epilepsies occurred with the same prevalence. Further, patients with seizure onset after age two were more likely to achieve seizure freedom. They also found that even in patients with drug-resistant epilepsy, seizures became less frequent as patients aged. There were no cases of sudden unexpected death in epilepsy or status epilepticus in their cohort, and over half (57%) had abnormal EEG backgrounds. Focal epileptiform discharges were seen in 76%, 19% had generalized epileptiform discharges, and only one patient (4%) had a normal EEG without epileptiform discharges[3].
Attempts to evaluate genotype-phenotype correlations have been inconclusive. Individuals with missense mutations may be more likely to develop seizures as compared to other types of mutations[2]. It has also been proposed that terminal variants occurring in exons 9-19 cause the full PTHS clinical phenotype, while variants which occur earlier in the gene may have a more variable or mild presentation[13]. However, there is a case report of an individual with a single base-pair deletion in exon 19 who has only mild intellectual disability, speaks in full sentences, and achieves motor milestones on time[6]. However, These studies are limited by small sample sizes, and in a relatively large case series, there was no clear correlation between disease severity and the type or location of the variant[1].
As in many other single-gene disorders, precision therapies are in development for PTHS, though only supportive care is available currently. With potential therapies on the horizon, clinical assessments that may track disease severity over time are needed. EEG is a potential biomarker that is often utilized in other neurodevelopmental disorders and genetic epilepsies. Systematic evaluation of EEGs in the PTHS population is very limited. In this study, we reviewed the available clinical EEGs and qualitative reports from our Pitt-Hopkins Syndrome Specialty Clinic to assess electrographic signatures that may be unique to PTHS, as demonstrated in other disorders, including Angelman syndrome[14].
METHODS
We conducted a retrospective chart review of the patients with PTHS seen in our Pitt-Hopkins Specialty Clinic at the University of Texas Southwestern and Children’s Health Dallas Center for Autism and Developmental Disabilities from 2016 to 2021. There are a total of 32 patients with PTHS in the database, and 16 had a clinical EEG report available to review. We reviewed clinical data, including demographics, neurological manifestations of PTHS, developmental abilities, brain imaging reports, and EEGs. This study was conducted in accordance with the Declaration of Helsinki and received institutional review board approval from the University of Texas Southwestern Medical Center.
Clinically obtained magnetic resonance imaging (MRI) of the brain was reviewed where available. The MRIs performed at our center were read and reported by board-certified pediatric neuroradiologists. For brain imaging performed at outside institutions, only reports were available for review. Imaging reports were classified as normal or abnormal. For abnormal studies, findings were categorized as microcephaly, gliosis, myelination abnormalities, hypoplasia or atrophy, and dysplasia of the corpus callosum.
EEGs were obtained for clinical indications. The most common clinical indication was clinical seizures or events concerning seizures. EEGs obtained at our institution were recorded using a video EEG machine (Stellate Harmonie, 2009-2011 and Natus Xltek NeuroWorks, 2011-2021). Electrodes were applied with paste utilizing the 10-20 international system. A single-lead electrocardiogram was included for all recordings. All EEG recordings performed at our center were read and reported by board-certified pediatric epileptologists. Some of the EEGs were performed at outside institutions, reports of these EEGs were reviewed, and results were analyzed. EEGs included in the study were either continuous long-term video EEGs (> 23 h) or routine (< 2 h) recordings. EEG recordings were sorted by type (long-term or routine), age of the patient at the time of the study, and normal or abnormal impression. If EEGs were abnormal, they were grouped into epileptiform abnormalities, background abnormalities, or both. EEG characteristics were analyzed in relation to the developmental profile, imaging findings, and epilepsy characteristics.
Descriptive statistics were used to describe the demographics of the cohort, genotype, clinical characteristics, and qualitative findings on brain imaging and EEG. Fisher’s exact test was used to compare rates of abnormal EEG, abnormal MRI brain, and delay of ambulation by greater than 1 year and greater than 3 years. This test was used because of the small sample size and the likelihood that there would be 5 or fewer observations in any given cell. The data analysis for this paper was generated using SAS software, Version 9.4 of the SAS System for the University of Texas Southwestern. Copyright © 2016 SAS Institute Inc. SAS and all other SAS Institute Inc. product or service names are registered trademarks or trademarks of SAS Institute Inc., Cary, NC, USA.
RESULTS
At the time of their initial EEG, patients ranged in age from 15 months to 9 years (mean 3.75 years, standard deviation 2.25 years). All individuals had developmental delay, five had epilepsy, and seven had breath-holding spells, as previously reported [Table 1][1]. We reviewed a total of 29 EEG reports, representing 16 unique individual patients. EEG recordings were available for 12 of the 29 EEGs, representing 6 of the 16 individual patients, and no additional abnormalities were identified beyond those identified in the clinical report. Of the EEG recordings reviewed, which were reported as normal, all parameters including posterior dominant rhythm, sleep-wake transitions, and sleep architecture were normal when accounting for age at the time of the EEG. Seventeen of the 29 EEGs reviewed were abnormal, and nine out of sixteen individual patients had at least one abnormal EEG (56%). Most EEGs were obtained to characterize seizure-like events. Median age at first EEG was 3.5 years, and among children with an abnormal EEG, median age at first abnormal EEG was 4.5 years. Abnormalities included interictal epileptiform discharges (4/9, 44%), generalized slowing (5/9, 56%), and focal slowing (4/9, 44%), [Table 2]. Three of the four individuals with epileptiform abnormalities had focal spikes only, and one patient had both multifocal and generalized spikes. Focal spikes were seen most often over the centrotemporal and occipital regions [Figure 1]. All four children who had epileptiform abnormalities also had background slowing: three (75%) had generalized slowing, and one (25%) had focal slowing. There were four patients (4/9, 44%) who had abnormal EEG with background slowing without epileptiform abnormalities. One patient with an initial normal EEG had subsequent abnormal EEG with focal slowing and focal spikes. Once a patient had an abnormal EEG, findings were consistently demonstrated on repeat studies. Of the five patients with epilepsy, all had focal onset seizures with or without impaired awareness, and some had secondary bilateral tonic-clonic seizures. All four individuals with interictal epileptiform discharges had clinical seizures, and one individual with clinical seizures had focal slowing on a routine EEG [Table 2]. There were two EEGs that captured typical hyperventilation spells with oxygen desaturation that did not have EEG correlate.
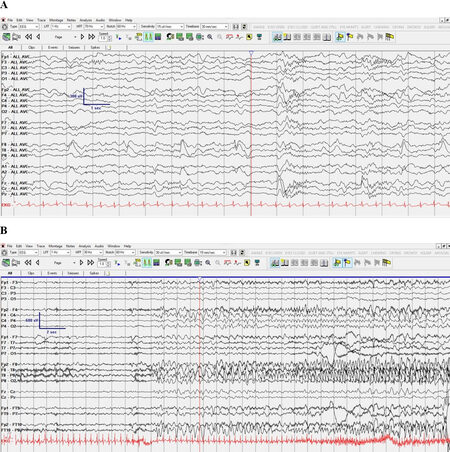
Figure 1. EEG demonstrating focal slowing, focal spikes, and a focal onset seizure in a patient with an intronic splice site missense variant (patient #18 in [Table 1]). EEG was performed at age 3 years. A: Right temporal delta slowing with right temporal spikes in common average reference montage; B: Right temporal onset seizure in longitudinal bipolar montage, shown in 15 mm/s time scale. EEG: electroencephalogram.
Demographics and Clinical Features
| Gender (M:F) | 11:5 |
| Genotype: | Deletions – 5 (3 large deletions involving multiple genes) Missense – 6 Nonsense – 3 Duplication – 1 |
| Age Range at first EEG (mean, SD) | 15 months to 9 years (3.75 years, SD 2.25 years) |
| Clinical Features | Developmental Delay – 16 Epilepsy – 5 Breath-Holding Spells – 7 |
| EEG Abnormalities | Epileptiform Discharges – 4 Focal Slowing – 5 Generalized Slowing – 5 |
| MRI Abnormalities | Normal : Abnormal – 4:9 Hypoplasia or Atrophy – 6 Corpus Callosal Dysplasia – 4 Gliosis – 2 Myelination Abnormalities – 2 Microcephaly – 1 |
Genotype, imaging, EEG, and clinical features
| Subject ID | Gender | Age at 1st EEG | Genotype | Epilepsy | Walking Delayed by ≥ 1 year | EEG | MRI brain |
| Large deletion | |||||||
| 02 | M | 2yr | 6.757 Mb del 18q21.2 (20 genes) | No | X | Normal | Normal |
| 03 | F | 16m | 4.2 Mb deletion of Ch 18q21.2 (14 genes) | No | n/a | Normal | n/a |
| 04 | M | 5yr | 20kb del 18q21.2 | Left eye deviation with GTC | X | Right central and left temporal SW, Generalized Slowing | ACC, MC, Frontal hypoplasia |
| 11 | M | 7yr | 94kb del 18q21.2 | Behavioral arrest with generalized stiffening | X | Right occipital, parietoccipital SW, Bilateral occipital SW | Normal |
| 17 | F | 4yr | 7.6Mb deletion 18q21.2 (Entire gene) | X | Normal | n/a | |
| Missense | |||||||
| 10 | M | 2yr | c.1738C > T (Exon 18) | X | Normal | Thin CC, delayed myelination, Frontal hypoplasia | |
| 12* | M | 9yr | c.1933delG (Exon 19) | Normal | Dysplastic CC | ||
| 13 | M | 23m | c.457 - 461del (Exon 12) | X | Background slowing | Diffuse atrophy, punctate gliosis | |
| 18 | F | 2yr | c.1650 - 2 A > G (Intron 17) | Left-sided stiffening and jerking with altered consciousness | X | Right temporal SW, Right temporal slowing | Thin CC# |
| 27 | F | 4yr | c.922 + 3G > T (Intron 11) | X | Disorganized, slow background | Inferior vermis hypoplasia | |
| 28 | M | 4yr | c.1739G > A (Exon 18) | Eyes roll back, generalized stiffening with left arm jerking and perioral cyanosis | X | Multifocal SW Right fronto-temporal SW Generalized SW, Generalized slowing | MC |
| 31 | M | 3yr | c.1739G > A (exon 18) | X | Normal | Diffuse atrophy, Thin CC | |
| Nonsense | |||||||
| 16 | M | 6yr | c.680 - 682 del (Exon 10) | X | Bifrontal slowing | Paucity of WM | |
| 29 | F | 6yr | c.605delC (Exon 9) | X | Right centro-temporal slowing | Delayed myelination# | |
| 30 | M | 15m | c.1169del (Exon 15) | Left-sided jerking with retained consciousness | n/a | No SW, Right centroparietal slowing | PV gliosis |
| Duplication | |||||||
| 05 | M | 2yr | 201 kb duplication of Ch 18q21.2 (Exons 5 - 8) | X | Normal | n/a | |
Brain MRIs were available in 13 of the 16 patients who had EEGs performed, and the majority were abnormal (9/13). Abnormalities included hypoplasia or atrophy of brain parenchyma (6/9), corpus callosal dysplasia (4/9), abnormalities of the white matter, or myelination patterns (2/9), gliosis (2/9), and microcephaly (1/9), [Table 2]. Of the four patients with a normal brain MRI, two patients had a prior study with abnormalities, one had delayed myelination, and the other had thinning of the corpus callosum.
Developmentally, all individuals demonstrated delays in the achievement of motor and language milestones. With regards to gross motor milestones, those with an abnormal EEG (n = 9) attained rolling at a median of 4.5 months (range 3 to 28 months), sitting at a median of 14 months (range 6 to 32 months), standing at a median of 36 months (range 24 to 36 months), cruising at a median of 39 months (range 30 to 48 months), and walking at a median of 48 months (range 15 to 108 months). Five of the nine were able to babble at a median age of 12 months (range 6 to 40 months), but none of the individuals had words at their most recent follow-up (age range 2 to 25 years). Those with abnormal EEGs attained motor milestones at similar ages as those with normal EEGs but are markedly delayed compared to the average age of motor skill acquisition in typically developing children [Figure 2].
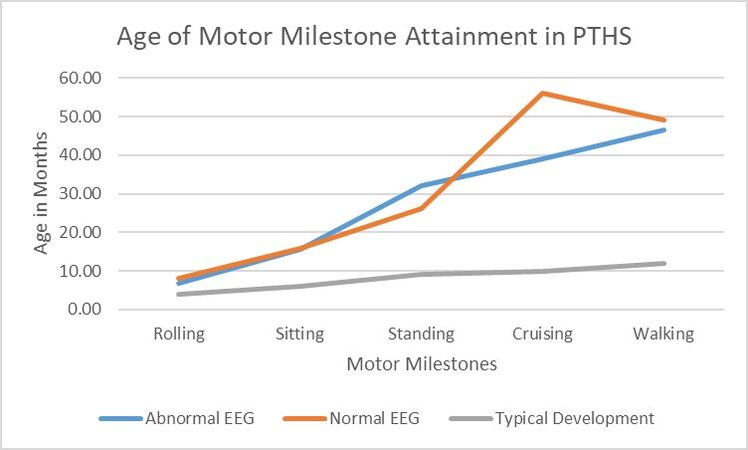
Figure 2. Age of Motor Milestone Attainment in PTHS. In this line graph, we demonstrate the mean age at which individuals with PTHS attain motor milestones of rolling, sitting, standing, cruising, and walking as grouped by those with abnormal EEG and those with normal EEG. The mean age of attainment in typically developing individuals is depicted for reference. PTHS: Pitt-Hopkins syndrome; EEG: electroencephalogram.
Fisher’s exact test was used to examine the relationship between EEG abnormalities and clinical features of PTHS, including abnormal MRI brain imaging and delayed ambulation. There was a higher rate of abnormal brain MRIs among patients with an abnormal EEG compared to patients with only normal EEGs (8/9 vs. 3/7, P = 0.03). When assessed individually, there was not a significant difference in the rates of abnormal MRI brain among patients with epileptiform discharges, generalized slowing, or focal slowing. To assess the relationship between developmental motor abilities and abnormal EEGs, we dichotomized delays in ambulation to at least 1-year delayed or at least 3-year delayed onset of ambulation. There was no significant relationship between abnormal EEGs and delayed ambulation of 1-year or greater, but there was a higher prevalence of abnormal EEGs among those patients with a 3-year or greater delay in ambulation (P = 0.2 vs. 0.03, respectively).
We also assessed genotype-phenotype correlations. Our cohort included a range of genotypes: large deletions (5/16), missense variants (6/16), nonsense variants (3/16), and one duplication. Because of prior studies demonstrating a higher prevalence of epilepsy in patients with missense variants, we evaluated the relationship between clinical severity in those with missense variants in comparison to non-missense variants. Rates of abnormal EEGs, abnormal brain MRIs, diagnosis of epilepsy, and delayed ambulation were similar between both groups. Delayed ambulation was a ubiquitous finding among all 16 individuals. Epilepsy was present in 2/5 with a large deletion, 2/7 with a missense variant, and 1/3 with a nonsense variant. Abnormal EEGs were present in 3/5 with a large deletion, 4/7 with a missense variant, and all 3 with a nonsense variant. The one patient with a duplication did not have epilepsy, and his EEG was normal[1].
DISCUSSION
This review of EEGs in patients with PTHS showed that there was no characteristic pattern, and approximately half of the patients had normal EEGs. In patients with abnormal EEGs, focal epileptiform abnormalities were more common than generalized, and none of the patients had only generalized spikes. In contrast to a recently published series of 21 patients with PTHS in which 4.8% of their cohort (1/21 patients) had no epileptiform discharges on prolonged EEG (3), we found a much higher proportion of normal EEGs (7/16, 43%). However, both studies support the finding that there is no characteristic signature EEG pattern in PTHS and focal epileptiform discharges with focal-onset seizures appear to be more common than generalized discharges and seizures[3]. This variance could be explained by varying duration (i.e., 1-h vs. 24-h or longer EEG) or age at the time of EEG between the two cohorts. A larger prospective study controlling for these variables is needed. There was no specific correlation between EEG results and developmental trajectories or age in our patient group.
All individuals with clinical epilepsy had focal onset seizures and abnormal EEGs. The majority had epileptiform discharges, and one had focal slowing. This suggests that in the presence of epileptiform EEG abnormalities, there is a high likelihood of seizures and epilepsy. In contrast, none of the individuals with normal EEGs were diagnosed with clinical epilepsy. Larger studies are needed to confirm this observation, which may guide the prognosis and diagnosis of epilepsy in PTHS. PTHS patients have three types of paroxysmal events: epileptic seizures, abnormal breathing spells, and motor stereotypies. Because patients can have any combination of these events, it can be challenging to clinically distinguish between spell types. Consequently, abnormal breathing spells are frequently misdiagnosed as epileptic seizures. Further, patients can have apnea or overbreathing just prior to onset of seizures which can add to the diagnostic challenge[4]. Breath-holding events were present in nearly half of our cohort, and these events, when captured on EEG, were not epileptic. Epilepsy monitoring unit evaluation with video EEG recording can be helpful for the characterization of spells and accurate diagnosis and management.
Previous studies report an increased frequency of epilepsy in patients with missense mutations; however, there has not been a reliable or reproducible genotype-phenotype correlation in this disorder[2]. We did not identify any specific phenotypic differences (e.g., diagnosis of epilepsy, abnormal EEG, abnormal MRI, delayed ambulation) among those with a missense variant compared to those with a non-missense variant. The TCF4 gene encodes for transcription factor 4, which is a basic helix-loop-helix E protein responsible for neuronal differentiation in early brain development. Haploinsufficiency of TCF4 has been associated with significant dysregulation of pathways critical for brain development and function, including synaptic and neuronal plasticity, calcium signaling, and neurotransmission through altered expression of neuroreceptors[15]. TCF4 may be transcribed into at least 18 different isoforms with varying N-termini, which impact subcellular localization and function[16]. Functional analyses and mapping of missense variants reveal that different functional domains exist within the TCF4 gene and can alter transcriptional activation of downstream genes, including NRXN1 and CNTNAP2, which cause Pitt-Hopkins-like syndromes 1 and 2, respectively[17]. The variable effects of pathogenic variants on TCF4 functioning likely contribute to phenotypic variability[18]. Larger studies of patients coupled with functional variant analyses are needed to fully explore the genotype-phenotype relationship in PTHS.
Clinically, patients with PTHS resemble those with Angelman and Rett syndromes, with severe developmental delays, limited to absent language abilities, motor incoordination, abnormal breathing spells, and epilepsy. Patients with Rett and Angelman syndromes tend to have abnormal EEG with recognizable patterns. In Rett syndrome, this is characterized by EEG that is initially normal, then abnormal with diffuse slowing, rhythmic theta slowing in the centrotemporal region, and multifocal and generalized epileptiform discharges[19]. In Angelman syndrome, the most common pattern consists of high amplitude frontal delta activity with superimposed epileptiform discharges[14]. In contrast, as we demonstrate here, patients with PTHS seem to lack a characteristic EEG signature and can have a normal EEG background for age. When they do have abnormalities on EEG, they differ between patients and can include generalized slowing, focal slowing, and focal epileptiform discharges. A normal or non-specific EEG with a clinical phenotype suggestive of Angelman or Rett syndrome should raise the consideration for a diagnosis of Pitt Hopkins syndrome.
There were three major limitations in this study: (1) small sample size; (2) EEG reports were generated by multiple different institutions; and (3) EEGs were obtained with a clinical suspicion of seizures. Because EEGs were completed at numerous institutions, reports did not contain a unified set of elements, and we were only able to review tracings on the EEGs that were completed at our institution. Therefore, abnormalities reported in the studies completed by other institutions could not be independently verified by a single EEG reader. Further, studies were not uniform in their duration, and included both routine EEGs and prolonged epilepsy monitoring unit studies. Finally, there was a selection bias because only clinically-obtained EEGs were reviewed. EEGs were obtained in this subpopulation of PTHS patients only when there was a clinical suspicion for seizure. This selection bias could lead to an over-representation of epilepsy in our cohort, or conversely, there may be a higher proportion of abnormal EEGs that exist in the total PTHS population even in the absence of clinical epilepsy. A prospective natural history study would help evaluate the spectrum of EEG abnormalities along with the evolution of EEG findings and epilepsy with age in PTHS by standardizing longitudinal data collection.
In conclusion, PTHS is a rare, genetic, neurodevelopmental disorder that presents with a homogenous phenotype of profound global developmental delay characterized by markedly delayed ambulation and most patients are unable to use spoken language to communicate. PTHS can have phenotypic similarities with Angelman syndrome and Rett syndrome; however, they lack a characteristic EEG signature. In a patient presenting with an Angelman-like phenotype and a normal or a non-characteristic EEG, a diagnosis of PTHS may be considered. Further, it is of note that breath-holding spells like those seen in Rett syndrome do not have an epileptic ictal EEG correlate.
Although a distinctive EEG signature was not identified, all individuals with epilepsy in our cohort had focal onset seizures with corresponding focal epileptiform discharges or focal slowing on EEG. Further research is needed to fully understand why a pathological variant in a transcription factor responsible for neuronal differentiation would yield focal onset seizures.
DECLARATIONS
AcknowledgmentsThe authors would like to acknowledge the families caring for individuals with Pitt-Hopkins syndrome who have trusted us with the medical care of their loved ones as well as the support of the UTSW Department of Pediatrics for support of clinical research in children with rare neurodevelopmental disorders.
Authors’ contributionsContributed to the conception of the study, data analysis, and drafting of the manuscript: Bone M, Goodspeed K, Sirsi D
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was funded in part by support of the UT Southwestern Dedman Family Clinical Scholar Endowment fund.
Conflicts of interestKG has received research support from Taysha Gene Therapies and Neurogene, Inc. for work that is unrelated to that presented in this manuscript. MB has no disclosures to report. DS has no relevant disclosures to report.
Ethical approval and consent to participateThis study was conducted in accordance with the Declaration of Helsinki and received institutional review board approval from the University of Texas Southwestern Medical Center.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Goodspeed K, Newsom C, Morris MA, Powell C, Evans P, Golla S. Pitt-Hopkins syndrome: a review of current literature, clinical approach, and 23-patient case series. J Child Neurol 2018;33:233-44.
2. Sweatt JD. Pitt-Hopkins syndrome: intellectual disability due to loss of TCF4-regulated gene transcription. Exp Mol Med 2013;45:e21.
3. Matricardi S, Bonanni P, Iapadre G, et al. Epilepsy, electroclinical features, and long-term outcomes in Pitt-Hopkins syndrome due to pathogenic variants in the TCF4 gene. Eur J Neurol 2022;29:19-25.
4. Zollino M, Zweier C, Van Balkom ID, et al. Diagnosis and management in Pitt-Hopkins syndrome: first international consensus statement. Clin Genet 2019;95:462-78.
5. Watkins A, Bissell S, Moss J, et al. Behavioural and psychological characteristics in Pitt-Hopkins syndrome: a comparison with Angelman and Cornelia de Lange syndromes. J Neurodev Disord 2019;11:24.
6. Tan A, Goodspeed K, Edgar VB. Pitt-Hopkins syndrome: a unique case study. J Int Neuropsychol Soc 2018;24:995-1002.
7. Currò A, Doddato G, Bruttini M, et al. CDKL5 mutations may mimic Pitt-Hopkins syndrome phenotype. Eur J Med Genet 2021;64:104102.
8. Pearson E, Watkins A, Oliver C, Karim A, Clayton-Smith J, Welham A. The adaptive functioning profile of Pitt-Hopkins syndrome. Eur J Med Genet 2021;64:104279.
9. Balkom ID, Vuijk PJ, Franssens M, Hoek HW, Hennekam RC. Development, cognition, and behaviour in Pitt-Hopkins syndrome. Dev Med Child Neurol 2012;54:925-31.
10. Carotenuto M, Roccella M, Pisani F, et al. Polysomnographic findings in fragile x syndrome children with EEG abnormalities. Behav Neurol 2019;2019:5202808.
11. Maini I, Cantalupo G, Turco EC, et al. Clinical and polygraphic improvement of breathing abnormalities after valproate in a case of Pitt-Hopkins syndrome. J Child Neurol 2012;27:1585-8.
12. Kirikae H, Uematsu M, Numata-Uematsu Y, et al. Two types of early epileptic encephalopathy in a Pitt-Hopkins syndrome patient with a novel TCF4 mutation. Brain Dev 2022;44:148-52.
13. Bedeschi MF, Marangi G, Calvello MR, et al. Impairment of different protein domains causes variable clinical presentation within Pitt-Hopkins syndrome and suggests intragenic molecular syndromology of TCF4. Eur J Med Genet 2017;60:565-71.
15. Kennedy AJ, Rahn EJ, Paulukaitis BS, et al. Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep 2016;16:2666-85.
16. Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5’ exon usage and splicing. PLoS One 2011;6:e22138.
17. Forrest M, Chapman RM, Doyle AM, Tinsley CL, Waite A, Blake DJ. Functional analysis of TCF4 missense mutations that cause Pitt-Hopkins syndrome. Hum Mutat 2012;33:1676-86.
18. Sepp M, Pruunsild P, Timmusk T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet 2012;21:2873-88.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Bone M, Goodspeed K, Sirsi D. Epilepsy and electroencephalography in Pitt-Hopkins syndrome. J Transl Genet Genom 2022;6:169-78. http://dx.doi.org/10.20517/jtgg.2021.56
AMA Style
Bone M, Goodspeed K, Sirsi D. Epilepsy and electroencephalography in Pitt-Hopkins syndrome. Journal of Translational Genetics and Genomics. 2022; 6(2): 169-78. http://dx.doi.org/10.20517/jtgg.2021.56
Chicago/Turabian Style
Bone, Megan, Kimberly Goodspeed, Deepa Sirsi. 2022. "Epilepsy and electroencephalography in Pitt-Hopkins syndrome" Journal of Translational Genetics and Genomics. 6, no.2: 169-78. http://dx.doi.org/10.20517/jtgg.2021.56
ACS Style
Bone, M.; Goodspeed K.; Sirsi D. Epilepsy and electroencephalography in Pitt-Hopkins syndrome. J. Transl. Genet. Genom. 2022, 6, 169-78. http://dx.doi.org/10.20517/jtgg.2021.56
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 14 clicks
Cite This Article 14 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.