
Transcriptomics insights into interpreting AMD-GWAS discoveries for biological and clinical applications

Abstract
Genome-wide association studies (GWAS) have been successful in identifying genetic risk factors for a large number of complex diseases, including age-related macular degeneration (AMD), which is a highly heritable complex disease affecting millions of elderly individuals. However, the progress of elucidating the functional relevance of genetic findings in AMD has been slow, as most risk factors are non-coding, and we have little insight into the causal genes and disease mechanisms. In the last few years, gene expression regulation is emerging as a dominant mechanism through which GWAS risk variants lead to the disease. The purpose of this review is to provide an overview of how transcriptome studies can help in identifying the genes, pathways and therapeutic targets underlying GWAS discoveries in AMD. These approaches help pave the road for mechanistic understanding of GWAS findings and drive translational advances that will lead to improved AMD management and treatment.
Keywords
INTRODUCTION
Understanding the genetic basis of common diseases remains a subject of significant interest and challenge in the post-genome era. Genome-wide association studies (GWASs) have been instrumental in bringing us closer to the goal of understanding the genetic basis of complex diseases faster than ever before. GWAS have been conducted in ~3000 diseases and quantitative traits that have led to the identification of 71,673 trait-associated single nucleotide polymorphisms (SNPs) from 5687 studies[1]. These studies have provided the following insights into the genetic architecture of complex diseases; (1) complex traits are highly polygenic; (2) multiple independent association signals at each locus are not uncommon; (3) effect sizes of individual associated variants are small; and (4) the majority (~93%) of disease- and trait-associated variants lie within the noncoding sequence.
GWASs have undoubtedly established the critical role of common variants in complex diseases. However, these findings did not immediately reveal the causal variant, gene or biological mechanisms underlying the observed genetic association. Thus, it is time to envision the post-GWAS phase of human genetics that focuses on accelerating progress in moving genetic association to functional interpretation. Translating the genetic findings to meaningful biology warrants having a comprehensive picture of the causal variants and genes, disease-relevant cells, tissues and organ through which genes act, and the biological pathways and mechanisms driving the disease. In the past few years, functional studies to connect GWAS to biology have been gaining momentum, which could be largely attributed to our enhanced ability to generate and integrate genomic data at the genome, transcriptome and epigenome level[2]. Gaining a molecular understanding of genetic findings warrants the integration of genetic results with other biological data types from disease-relevant tissues and cell types and careful functional studies that ought to be tailored for different diseases.
Age-related macular degeneration (AMD) is the leading cause of irreversible vision loss resulting from the death of light-sensing photoreceptors, primarily in the central region of the retina called the macula. It is a complex multifactorial disease caused by the cumulative impact of genetic predisposition, environmental stress and advanced aging[3-5]. AMD afflicts almost 10 million individuals in the United States alone[6]. Fundus features such as large, soft drusen and pigmentary abnormalities are associated with an increased risk of AMD progression[7]. It is a progressive disease with early, intermediate and late stages of disease. Early/intermediate AMD is the most common and least severe form, characterized by pigmentary abnormalities in the macula and accumulation of extracellular aggregates of proteins, lipids and cellular components (called drusen). AMD in the advanced stage is subdivided into dry (geographic atrophy, or GA) and wet (choroidal neovascularization, or CNV)[8]. In the United States, over 1.75 million people have advanced stage of AMD, and 7.3 million people are affected with intermediate stage and are at the risk of developing advanced AMD. Patients with neovascular AMD respond well to treatment with anti-vascular endothelial growth factor (VEGF) agents, but those diagnosed with the dry form of the disease (approximately 85% of AMD cases) have no approved treatment options available to them[9]. Detecting the disease early can provide a window of opportunity to avoid or delay the burden of vision loss on both the patient and their families. The earlier that AMD is detected, the earlier steps can be taken to help slow its progression and save sight through treatment and/or lifestyle modifications. However, there is a paucity of studies on the early and intermediate stages of AMD, and as a result, there are no reliable biomarkers for predicting the disease progression to either GA or CNV. In the absence of treatment, antioxidant vitamins and mineral supplements remain the best option for slowing the progression of the disease in individuals at a high risk of developing AMD[10].
GENETICS STUDIES IN AMD
Remarkable progress has been made in delineating the genetic basis of AMD. Familial aggregation, twin studies, and segregation analyses indicated substantial genetic contributions to AMD pathogenesis that encouraged researchers to identify genetic risk factors associated with AMD through candidate gene, linkage, and association studies. Traditional linkage mapping studies in large and small families with AMD were considerably successful in identifying the genetic loci at chromosome 1q1q31, 9p13, 10q26, and 17q25[11-13]. At the same time, human genome sequencing and development in methods to genotype genome-wide variants in humans led to the transition to GWAS for genetic studies in AMD. The first GWAS using merely 50 controls and 96 cases was successful in uncovering the strong association of a coding variant (Y402H) in complement factor H (CFH) with AMD[14-16]. Subsequent studies have identified additional non-coding variants at CFH, in other complement genes, and at the ARMS2/HTRA1 locus (reviewed in Ref.[17]). In recent years, two GWAS with a large number of cases and controls have been performed. An international collaborative effort on the meta-analysis of AMD-GWAS from 18 centers, involving 17,000 AMD cases and 60,000 matched controls of European and Asian ancestry, reported 19 loci[18]. The most recent AMD-GWAS was performed by International AMD Genomics Consortium, which surveyed genome-wide variants to evaluate 16,144 patients with advanced AMD and 17,832 controls and identified 52 independent common and rare variants at 34 loci that exhibited an association with AMD, accounting for more than half of genomic heritability and suggest the involvement of complement, lipid metabolism/cholesterol transport, angiogenesis, and extracellular matrix reorganization pathways in AMD pathology[19,20].
Although AMD is a progressive disease, majority of the genomic studies have focused on the late-stage disease that limits our understanding of molecular and genetic underpinnings in early and intermediate stages. More recently, two large-scale studies have looked into the genetic basis of early and intermediate AMD. Fritsche et al.[19] tested the association of 34 known AMD in 6657 cases of intermediate AMD and reported a substantial overlap of genetic determinants (24/34 known AMD risk variants) between advanced and intermediate AMD. However, 10 risk variants showed no association with intermediate AMD despite sufficient statistical power to detect the association[19]. Another GWAS meta-analysis of 11 data sources, including 14,034 early AMD and 91,214 controls, identified 10 loci with 8 overlapping with advanced AMD loci and two novel ones[21]. These studies emphasize the importance of large-scale population-based studies to gain insights into the genetic factors contributing to AMD progression.
Contribution of rare variants in AMD
The role of both common and rare variants contributing to the genetic architecture of complex traits has long been recognized[22]. It has been suggested that rare variants might contribute to the heritability in AMD[19,23]. Targeted sequencing of candidate genes and whole-exome sequencing identified rare coding variants in the CFH[24], C3[25-27], CFI[26,28], C9[26], CFB[29] and COL8A1[30] genes within GWAS loci. Additional studies also corroborated the findings of rare variants in complement genes[31-36], establishing an unequivocal role of the complement system in AMD pathology. So far, rare variants have been identified for AMD only in a limited number of case-control studies using exome chips[19], exome sequencing[30,37], and/or whole-genome sequencing[36]. In addition, exome sequencing in large, multiplex families also confirmed the occurrence of rare variants in the CFH, CFI, C9 and C3 genes at multiple AMD loci[27,34,35,38-42]. However, most of these studies were confined to looking at the rare variants within known AMD loci, and the rare variants outside of the GWAS loci remain relatively unexplored. A recent study performed exome sequencing of 264 individuals from 63 multiplex families with AMD and reported rare variants within eight AMD-GWAS loci and 13 genes outside the AMD-GWAS loci[43]. Further independent replications and molecular investigations of rare variants are needed to understand their role in AMD pathogenesis.
STEPS TO CONNECT GWAS DISCOVERY TO BIOLOGY
GWAS unquestionably established a strong genetic component of AMD and provided a broad framework for elucidating the contribution of genetic variants to AMD. It allowed a hypotheses-free discovery of disease variants that revealed novel disease biology and provided a wealth of clues for biological experimentation, risk prediction, disease progression, and biomarker and therapeutic target discovery [Figure 1]. However, we must also acknowledge that progress in translating genetic findings into understanding disease mechanisms and new cures and treatments for AMD has been slow to arrive. There are no biomarkers for predicting the disease progression to either GA or CNV. Unfortunately, targeting GWAS implicated pathways has not been effective in preventing/slowing disease progression or treatment as many clinical trials that relied on these hypotheses have failed to ameliorate AMD[44].
Figure 1. Applications of AMD-GWAS findings in understanding the disease biology and clinical practices and interventions. AMD: Age-related macular degeneration; GWAS: genome-wide association studies.
Drug targets with human genetic evidence of disease association are twice as likely to succeed as approved drugs[45]. Thus, investments in the functional follow-up of genomic findings are likely to be beneficial for AMD drug development. However, several factors have made it difficult to bridge the gap between the statistical associations of genetic variations and a functional understanding of the biology underlying AMD risk. First, GWAS variants are not causal. The causative SNP may lie anywhere within the linkage disequilibrium (LD) block surrounding the associated SNP that can span over 100 kb and often contain over a thousand individual SNPs. Second, a majority of associated variants reside in non-coding regions of the genome, thus underlying causative genes or pathological mechanisms are not immediately obvious. Some of the AMD-specific challenges are also worth noting. For example, modeling of AMD presents a substantial challenge[46]. AMD causes vision loss because of degeneration of photoreceptors in the central region of the retina called the macula, which is a primate-specific structure shared only by humans, monkeys and apes[47]. Thus, mouse models fall short of recapitulating many aspects of AMD. Similarly, there is a dearth of cell lines for ocular studies, and transformed cell lines such as ARPE-19 do not express key phenotypic features of human RPE[48]. Finally, we have a limited understanding of molecular and genetic underpinnings in the early and intermediate stages of the disease.
There are four key steps in elucidating the disease mechanisms underlying AMD risk variants: (1) identification of causal variant(s) and target gene(s) at the associated loci; (2) annotating variants and genes at tissues and single-cell resolution; (3) identifying molecular and cellular pathways associated with the disease; and (4) developing appropriate disease models to infer disease mechanisms. As a vast majority of GWAS variants fall into non-coding regions and often overlap enhancers, promoters and open-chromatin regions, gene expression regulation is emerging as a dominant mechanism in mediating disease risk. Thus, here we will be focusing on various ways gene expression studies can be leveraged to understand the risk variant-mediated disease causation.
TRANSCRIPTOME STUDIES IN DISEASE-RELEVANT TISSUES AND CELL TYPES
GWAS have established that > 90% of the risk variants associated with complex diseases including AMD reside in the non-coding regions that are not likely to directly affect the coding region of the gene. This brought attention to the annotation of the non-coding genome to understand its function. Enrichment of GWAS variants within regulatory DNA marked by deoxyribonuclease I (DNase I) hypersensitive sites (DHSs) led the focus on studying the gene expression regulation in the context of GWAS variants[49]. Additionally, these risk variants often disrupt binding sites for transcription factors (TFs), and these variable TF-interaction are believed to be the primary driver of phenotypic variation[50]. Additionally, cell context is a key determinant of gene regulation. Thus, a comprehensive understanding of global transcriptome regulation in disease-relevant cells and tissues represents the first logical step in functional understanding of GWAS findings. Towards this goal, Genotype-Tissue Expression (GTEx) project was initiated to establish a large resource for studying the relationship between genetic variations and gene expression levels across multiple tissues and cell types[51]. Unfortunately, the exclusion or limited representation of AMD-relevant tissues (retina, macula and RPE) has delayed the functional understanding of most ocular phenotypes. However, researchers in vision field were quick in recognizing this unmet need and have created several excellent resources in the last few years[52-59].
While tissue-shared regulation appears to underlie an appreciable proportion of the genetic component of complex traits[51,60], a series of studies have identified enrichment of GWAS signal in tissue-specific[60,61] or cell-type specific[62,63] expression quantitative trait loci (eQTLs). A recent single-cell sequencing study shows cell-type specificity for multiple AMD risk genes across diverse cell types, including bipolar cells (CFH, CFHR1), horizontal cells (HTRA1 and ARMS2), astrocytes (APOE) and microglia (CXC3R1)[57]. Thus, aligning cell-type-specific expression profiles with GWAS results can lead us one step closer to functional follow-up experiments[62,64]. However, human primary tissue samples are often a mixture of multiple cell-types, and tissue-specific expression is a function of the distribution of cell-types present in that tissue. A growing number of in silico deconvolution methods and associated reference panels with cell-type-specific marker genes enable the robust estimation of the enrichment of specific cell-types from bulk tissue gene expression data[65-67]. In recent years, single-cell sequencing technology has also emerged as key tool to gain insights into complex cell-types directly[68-71]. A comparative analysis of normal and disease conditions can help in characterizing complex cellular changes in response to pathology[72,73].
METHODS OF GENE EXPRESSION MEASUREMENT
Advent in next-generation sequencing has led to major advances in methods for gene expression profiling. In the last few years, RNA-sequencing (RNA-seq) has become the most common method of transcriptome profiling because of precise, quantitative measurement. Broadly, RNA-seq experiments can be divided into two categories:
1. Bulk RNA-seq: This method measures the gene expression from heterogeneous tissues for detection of a wide variety of RNA species, including mRNA, non-coding RNA, transcript isoforms, and circulating and small regulatory RNA[74]. It is relatively fast and inexpensive, and in the past decade, it has been widely used to investigate multiple aspects of biology and biomedical research including connecting GWAS findings to function[75]. Specifically, it has greatly facilitated in characterizing the molecular function of the human genome and the impact of genetic variation on gene expression levels[51]. Additionally, profiling of transcriptomes in large cohorts of cases and controls also presents an opportunity for identifying genes and pathways affected by the disease, enabling mechanistic insights into disease. However, as tissue is a mix of various cell-types of varying proportions, the bulk RNA-seq averages the expression across all cells in a sample, which can significantly confound the analysis of differential gene expression and eQTL analyses.
2. Single-cell RNA sequencing (scRNA-seq): This method allows the transcriptome profiling at the single-cell level, which can address many technical and biological limitations associated with bulk RNA-seq discussed above. This has great potential for providing insights into the understanding of biological diversity and rare cell-types that could not be resolved using bulk RNA-seq. scRNA-seq has been extensively applied for cell-types identification, classification and lineage tracing[76]. Cell-type-specific transcriptome, combined with variant information also offers an opportunity to implicate cell-type specificity of traits and diseases. In addition, this approach increases the power to detect such associations as expression profile is less heterogeneous for single-cell than bulk tissues. However, scRNA-seq resolution is often noisy, sparse, and high-dimensional, creating challenges for computational analysis. Additionally, it is still relatively expensive to apply to a large cohort for gaining insights into the cell types affected by the disease.
LEVERAGING TRANSCRIPTOMICS FOR IDENTIFICATION OF CAUSAL VARIANT AND TARGET GENE
Linking the AMD-GWAS variants to their target genes is an important step in understanding the role of the non-coding genome in disease. This can be done in two ways: firstly, by statistical fine-mapping through conditional association analysis that can help identify the credible variants that are most likely to be causal, and variants can be linked to causal genes through functional analysis[77]. However, identifying the causal SNP is complicated because of LD. Alternatively, GWAS loci can be analyzed for eQTLs[78,79] mapping where genotypes are correlated with the gene expression. They are divided into two classes: cis-eQTLs and trans-eQTLs [Figure 2]. eQTLs that affect the expression of transcripts from nearby genes (generally within 1 MB of transcription start site) are referred to as cis-eQTLs. These variants often regulate gene expression by influencing the transcription factor binding sites that can impact multiple levels of gene expression and chromatin organization such as histone modification and DNA methylation. cis-eQTLs are abundant in all species including humans. Across all tissues, 94.7% of all protein-coding and 67.3% of all long-non coding RNA (lincRNA) genes have an eQTL[51], whereas, in the retina, 81% of known protein-coding and 13% of non-coding genes are under genetic regulation[80]. Trans-eQTLs affect the expression of distant genes either on the same chromosome or elsewhere in the genome. Not much is known regarding the effects of trans-eQTLs in humans as they have a small effect size and a large number of samples need to be analyzed to discover trans effects. They are also far more variable across species and tissue-specific than cis-eQTLs[81].
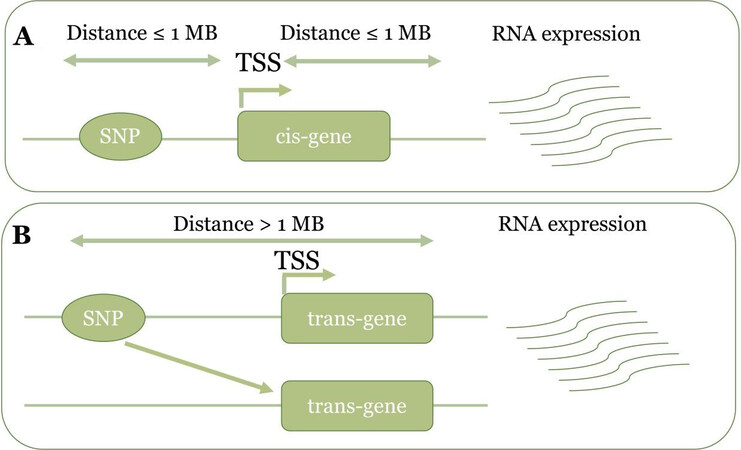
Figure 2. Gene expression regulation by a genetic variant. (A) cis-eQTL affects the expression of transcripts from nearby genes [generally within ± 1 MB of transcription start site (TSS)]. (B) trans-eQTL affects the expression of the distant gene ( > 1 MB) either on the same chromosome or elsewhere in the genome. eQTL: Expression quantitative trait loci.
Several lines of evidence suggest that a trait-associated variant is more likely to be associated with cis-eQTLs[78,82,83]. Thus, most analyses of integrating GWAS findings to gene expression regulation have focused on cis-eQTLs to identify the causal variant and target gene (gene whose expression is affected as a result of risk variant) [Figure 3]. In the early days, if a GWAS variant was also significant eQTL, it was considered to be causal. However, these analyses had a high number of false-positive results as genetic architecture underlying GWAS and eQTL signals were not accounted for. Newer methods integrate eQTL with the GWAS data to identify genes regulated by GWAS loci using colocalization methods[2], which apply probabilistic analysis in the Bayesian framework for SNP-level colocalization analysis that can provide molecular insights into complex diseases[84].
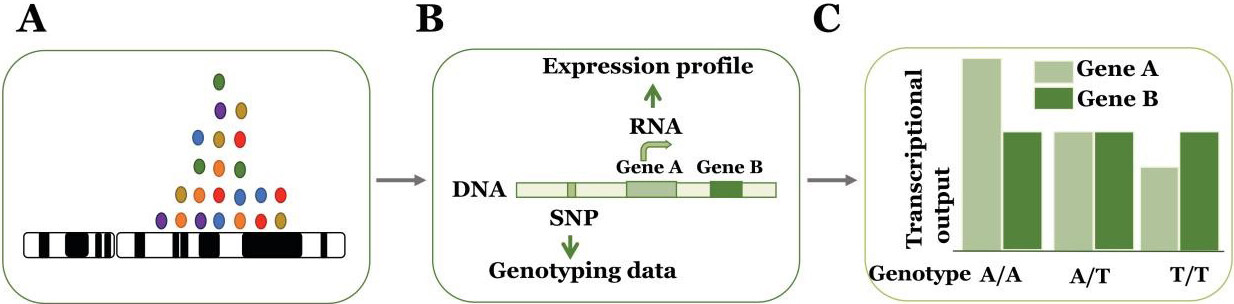
Figure 3. Identifying the causal variant and target genes using eQTL analysis. (A) GWAS variants are non-coding, and causal and non-causal disease variants are in strong LD. (B) eQTL analysis can identify the variants that affect gene expression in disease-relevant tissues. (C) A difference in expression (transcription) of gene A is mediated by genotypes (A/A, A/T/, T/T) leading to a phenotype. The expression of the gene B is not influenced by the genotypes. eQTL: Expression quantitative trait loci; GWAS: genome-wide association studies; LD: linkage disequilibrium; SNP: single nucleotide polymorphism.
eQTL STUDIES IN AMD
Ratnapriya et al.[80] reported the first reference eQTL map of over 500 post-mortem human donor retinae (from controls and AMD cases at early, intermediate or advanced stages) and reported 14,565 genetic variants (eVariants) that control the expression of 10,474 genes (eGenes). The integration of AMD-GWAS data with eQTLs showed that nine lead SNPs at the GWAS loci were significant eQTLs in the retina for 19 SNP-gene associations, and an integrative analysis highlighted the most plausible target genes at six AMD loci (B3GLCT, BLOC1S1, SH2B3, PLA2G12A, PILRB and POLDIP2/TMEM199)[80]. This data provided a basic framework for functional genomic dissection of AMD risk loci and other related ocular traits such as glaucoma and diabetic retinopathy[80]. A second study analyzed the gene expression in healthy retina (n = 311) that included the control retina data from the previous study[80], and reported 403,151 significant eQTL variants (eVariants) that regulate 3007 genes (eGenes)[85]. The integrated results from 16 published GWAS investigating 12 distinct ocular traits including AMD reported 65 unique GWAS variants to be regulating gene expression in retinal tissue[85]. Another study comprised of cis-eQTL analysis in human RPE/choroid and retina in bulk tissue samples from the macula and non-macular regions in ~120 donors (98 control and 23 AMD)[86]. They used colocalization of the GWAS and eQTL genetic signals and identified 15 putative causal genes at 13 known AMD loci (PILRA, PILRB, BAIAP2L2, TSPAN10, B3GLCT, TRPM1, SLC12A5-AS1, BLOC1S1, RDH5, TMEM199, BCAR1, COL4A3, TNFRSF10A, HTRA1 and CFI). Another study looked at the eQTL generated from 23 primary human fetal RPE lines under two metabolic conditions and reported much fewer eQTLs (687 shared, 264 glucose-specific, and 166 galactose-specific eQTLs) and colocalization analysis implicated four genes (RDH5, PARP12, WDR5 and EPB41L3)[87].
These studies suggest that variation in gene expression levels is widespread, highly heritable, and amenable to genetic mapping. The strongest eQTLs are found near the target genes, and there is a significant enrichment of AMD-associated variants in eQTLs. Tissue shared regulation is more prevalent than tissue-specific regulation; however, integrating the GWAS with eQTL in the disease-relevant tissues can provide key insights into the functional interpretation of AMD-GWAS loci[80]. eQTL data in the retina has been successful in identifying the genes at few AMD-GWAS loci, but many others remain uncharacterized. This could be attributed to the absence of well-powered eQTL data from RPE and choroid as well as relatively high experimental noise in the current eQTL studies. Future studies involving single-cell technology for molecular QTL mapping and including additional molecular phenotypes such as methylation and histone modification is likely to improve these studies.
LEVERAGING GENE EXPRESSION AND REGULATORY NETWORKS FOR DISCOVERING AMD GENES
GWAS revealed that multiple variants contributed to the risk of AMD. Additionally, identification of common risk variants in multiple genes from complement pathways indicated its involvement in AMD pathogenesis. These observations prompted studies for translating GWAS findings to the biological pathways that could reveal the series of events that culminates in the disease onset and explains the disease mechanism. Pathway exploration can be done in two ways: using the GWAS data or using molecular phenotypes such as gene expression measurements.
Pathway analysis in GWAS data
Traditional GWAS approach has the limitation of testing a single variant association with the trait of interest and conventional use of overly conservative multiple testing strategies with a genome-wide significance threshold of P < 5 × 10-8. An alternate approach to organizing summary statistics for these variants into biologically meaningful groups is to look at the overall effects of minor perturbations to genes and pathways. Pathways are defined by curated pathway databases or a collection of predefined gene sets for pathways based on prior biological knowledge, and the significance of each pathway can be summarized based on the disease association of markers in or near genes[88]. A recent analysis of AMD-GWAS data using this approach reported that eight genes (C2, C3, LIPC, MICA, NOTCH4, PLCG2, PPARA, and RAD51B) strongly contributed to significant pathways driving AMD association[89].
Differential expression analyses
A second approach to gain insights into the disease pathways is to compare the gene expression between normal and disease tissues. These differential expression (DE) analyses facilitate the identification of novel biological processes and genes involved in disease[90,91]. This approach overcomes the limitation of analyzing genes driven by genetic association and could reveal candidates that can offer an explanation for “missing heritability”. In AMD, RPE/choroid and the photoreceptors of the neural retina have been the primary focus of gene expression studies[92]. Analysis of these tissues from both macular and extramacular regions can offer key insights. However, the limited availability of these tissues, along with the technical and logistic difficulty of obtaining high-quality RNA from the postmortem tissues, have been rate-limiting steps in large-scale comparisons between normal and AMD donors. Multiple studies have looked into the gene expression changes during AMD and have reported several differentially expressed genes[80,92-94]. However, these findings have been difficult to replicate across studies, which could be attributed to several factors. First, variations in gene expression exist widely. Thus, using only small samples can lead to gene discovery that is reflective of interindividual as well as disease-associated changes. DE analyses also fail to capture small changes in gene expression because of other confounding factors such as age, gender and allele frequencies of GWAS risk variants between cases and controls. Cellular heterogeneity in the bulk RNA-seq adds another layer of complication in the interpretation of DE genes in the context of the disease. Incorporation of deconvolution methods and scRNA-seq in future comparative studies of normal and disease conditions can help in characterizing complex cellular changes in response to pathology[72,73].
Co-expression network analysis
Cellular processes are driven by multiple interacting genes and one might expect that genes whose expression is highly correlated will also have a functional correlation. Generation of such co-expression networks from the transcriptome data[95] have been used to associate genes of unknown function with biological processes, to prioritize candidate disease genes at the GWAS loci[96] or to pinpoint transcriptional regulatory modules in disease[97,98]. Weighted gene co-expression network analysis (WGCNA) is a widely used method to identify co-expressed gene modules[99]. Analysis of co-expression WGCNA modules built using human retina transcriptome data was shown to be enriched for genes within known AMD loci, and pathways involved in AMD (complement, extracellular matrix, and angiogenesis pathways) were closely connected[80]. These analyses can be further refined to dissect regulatory networks that are altered in disease and reveal genes that are likely to be regulators of disease processes. To further refine the modules and assign causality, genes within the module can be subjected to regulatory network construction. While these approaches may still leave a considerable number of candidates that may not be feasible for follow-up studies, integration of this approach with additional GWAS, eQTL and epigenomics datasets will aid in identifying disease AMD genes that remain unidentified through traditional genetic approaches.
Transcriptome-wide association studies
Gene discovery in GWAS is reaching saturation as the bulk of the remaining heritability is likely to be attributed to large numbers of common variants of small effects, rare variants, epigenetic changes, or environmental cues. Identification of novel associations with small effects requires large GWAS cohort sizes and lowering the genome-wide significance can lead to a higher false-positive rate. There has been a lot of progress on the methodologic approaches that integrate multiple data types, as they can offer unique insights and advantages in the interpretability of GWAS findings. Integrating gene expression with the GWAS can help in increasing the power of association as well as provide functional interpretation of GWAS findings. There are several methods for predicting the association between gene expression and the trait that leverages on eQTL reference, individual or summary-level GWAS data, and LD information. Transcriptome-Wide Association Studies (TWASs)[100] and Summary-data based Mendelian Randomization (SMR)[101] are two of the most commonly used methods for achieving this goal.
TWAS is a test for significant association between the cis component of gene expression and the GWAS trait that can help in identifying the target genes[100]. To date, two studies have looked into the association between the genetic component of gene expression and AMD. We showed that integrating GWAS summary level data[19] with eQTL generated for ~500 retinas led to the identification of 61 transcriptome-wide significant gene-AMD associations. Of these, 38 genes were present within 1 Mb of 14 AMD-GWAS loci and 23 genes outside the GWAS loci. This analysis helped in locating the target genes at the known loci as well as identifying RLBP1, PARP12 and HIC1 as likely new AMD-associated genes[80]. The second study analyzed the same GWAS data[19] with eQTL information from 27 different human tissues and reported 106 genes significantly associated with AMD variants in at least one tissue. Among these, 54 genes were significantly AMD-associated in one or more tissues and 16 genes (ADAM19, ARMS2, BTBD16, CFH, CFHR1, CFHR3, GPR108, PILRA, PILRB, PLA2G12A, PLEKHA1, PMS2P1, PPIL3, RDH5, STAG3L5P, and TNFRSF10A) were associated with AMD disease status in over 10 tissues[85]. These studies highlight the application of integrative analysis in biological insights.
MOVING FROM TISSUE TO CELL LEVEL RESOLUTION
The majority of approaches for identifying genes and pathways in complex diseases including AMD have used transcriptome profiles from the bulk tissues. However, tissues are made from many different cell types and bulk expression levels represent the average signals from multiple cell types present in that tissue. As discussed above, scRNA-seq presents a good alternative moving forward. However, currently we have limited single-cell data available for ocular tissues. Thus, the majority of studies have focused on identifying the cell types that are relevant in AMD pathology by looking at individual gene expression[57,86,102] or by performing cell-type-specific enrichment of AMD-associated genes[103]. Going onward, single-cell eQTL analysis will be critical for defining the precise cellular context for disease-associated risk variants and their target genes[104]. However, single-cell eQTL analysis has less discovery power, and profiling in a large cohort needed for such studies is still cost-prohibitive. Thus, a combination of bulk and single-cell analyses combined with deconvolution approaches can help in understanding the cellular context for disease-associated variants and their target genes. Finally, the construction of gene-regulatory networks at the single-cell level can help in unraveling the cell-type-specificity of gene interactions that leads to the disease.
BIOMARKER DISCOVERY IN AMD
A biomarker can be a substance or structure measured in body parts, fluids, or products that can affect or predict disease incidence, enable early detection of disease, and improve diagnostic classification to better inform individualized treatment. There have been several efforts to profile AMD-specific genomic, proteomic, metabolomic, and imaging-based biomarkers. Among these, studies involving systemic and ocular fluids (urine, tears, serum, and plasma) have achieved moderate success and implicated several components of the immune and complement system as potential biomarkers for AMD. Additionally, imaging-based structural and functional biomarkers have also shown great potential in predicting disease progression[105,106] and treatment outcomes[107]. They have the added advantage of being less invasive. Several excellent and comprehensive overview of all potential biomarkers and their applicability in AMD has been published[108-110]. Thus, here we focus our discussion on genomic biomarkers, which are measurable DNA and/or RNA characteristics that can be used as an indicator of normal biologic processes, pathogenic processes, and/or response to therapeutic or other interventions. These characteristics could be DNA alterations (single nucleotide polymorphisms, deletions or insertions, haplotypes, copy number variations) or RNA modification, whereas RNA characteristics can include RNA sequences, expression levels, RNA processing, and circulating RNA such as micro RNAs (miRNAs) and long-noncoding RNAs (lncRNAs).
The main aim of GWAS is biological discovery and gaining insights into the genes and pathways involved in AMD. However, in the light of immense success on AMD-GWAS, the development of DNA biomarkers holds great promises for personalized risk prediction and early detection. Several studies have looked into the utility of risk variants in AMD risk prediction and disease progression with varying success (reviewed in Refs.[111,112]). These could be further improved by integrating information from all risk variants, rare variants as well as molecular risk factors. Additionally, as AMD risk is conferred by multiple genetic variants, accessing the cumulative impact of risk allelesthrough polygenic risk scores (PRS) is likely to have better success for risk prediction[113]. PRS are the weighted sum of the individual effects of many risk alleles, with the weight based on the effect size estimated from GWAS and optionally re-estimated to account for other properties such as LD. Using data from two large clinical trials, Age-Related Eye Disease Study (AREDS) and AREDS2, it was shown that the addition of PRS to the demographic/environmental risk factors considerably improved the prediction performance[114]. A more recent study combining deep learning with survival analysis achieved high prognostic accuracy in predictions of progression to late AMD[115,116].
Despite analytical and clinical validation, a biomarker should demonstrate a clear effect in improving patient management, and an added value to the available treatment and management options for decision-making for AMD patients. However, at this stage gene therapy is not available for AMD and no direct benefits for patients have been demonstrated by identifying genetic risk factors. Thus, the American Academy of Ophthalmology currently does not recommend genetic testing for AMD patients. As future studies will shed more light on our understanding of disease etiology associated with AMD risk variants that show the value of treatment tailored to individuals’ genetic risk, genetic testing for AMD may become a routine in clinical practices.
There are even fewer examples of developing prognostic and predictive biomarkers based on gene expression signature. Several issues continue to impede progress in the clinic. One key limitation is the lack of transcriptome profiles available for AMD samples. Secondly, even when such studies are done, the sample sizes are small and a little overlap is reported when comparing gene signatures from different studies. Most studies also use convenience samples of heterogeneous patients for obtaining gene expression signatures, which makes it harder to replicate. A special class of RNA, called microRNA, is a small non-coding RNA molecule, thatis secreted in the circulation and exists in a stable form. As they are involved in post-transcriptional gene regulation, they have been good candidates for AMD biomarker discovery in systemic and ocular fluids. Multiple studies have performed comparative analyses of controls and AMD biofluids in search of biomarkers and have shown exciting possibilities, but the considerable inconsistency between different studies remains a major challenge for their clinical usefulness (reviewed Ref.[110]).
Biomarker identification requires a greater understanding of disease pathobiology. Thus, performing the transcriptome studies in the AMD-relevant tissues (RPE, retina and choroid) are likely to aid in biomarker discovery. However, gene signatures developed on these tissues can have limited application because of their inability to screen patients noninvasively. Thus, accessing the predictive capability of genes identified through genomic and molecular basis in circulating systemic and ocular fluids can have greater clinical potential. For example, VEGF and components of the complement system have been extensively investigated in AMD patients using metabolomic and proteomic approaches. Analysis of VEGF and its receptor in aqueous humor and plasma in various studies have failed to find a consensus change in VEGF level in AMD patients when compared to controls[110]. Similarly, the identification of complement proteins in histopathological and genetic studies prompted several comparative studies of complement regulators, complements components, and activation products in serums and plasma of AMD cases and controls (reviewed in Ref.[110]). Despite considerable inter-individual variability in complement level, few complement genes have shown a reproducible change in AMD cases, especially when integrated with AMD-associated common and rare variants in these genes[117]. This further enforces the value of genetic-data-driven biomarker discovery in AMD.
Whilst the potential of gene expression signatures remains largely unfulfilled, transcriptome studies in AMD hold great promises for interrogation of the biomarkers that could lead to novel insights into the underlying molecular processes. The increased technical advances in gene expression profiling and analysis including deep learning methods provide hope that signatures can begin to progress more frequently beyond the development phase and translate to patient benefit by identification of AMD biomarkers with predictive accuracy compared to more established prognostic factors.
CONCLUDING REMARKS AND FUTURE PERSPECTIVE
Elucidating the disease mechanisms underlying AMD-GWAS loci holds great potential [Figure 4]. In this review, we have focused mainly on the application of transcriptome data. However, with the increased availability of different data types such as histone modification, methylome, and proteome data, it is likely to improve and refine gene regulatory networks that get disrupted in AMD[20,118,119]. Additionally, understanding disease biology has the potential for developing an informed drug development program. Though still in infancy, the promises of genetics-based risk assessment in clinics are likely to become a reality in coming years. With a better understanding of causal variants underlying GWAS loci, polygenic risk scores will likely be incorporated as clinically useful predictive models of AMD. These advancements are key for developing reliable paradigms that make it possible to go from maps to mechanisms to medicine for AMD.
Figure 4. Applications of transcriptome data generated in control and AMD donors. Transcriptome data alone can help in identifying genes and pathways that are dysregulated in the disease process. They can provide insights into the disease biology as well as possibly identify disease biomarkers and therapeutic targets. Integrative analysis of transcriptome with the genetic data can be applied for eQTL and transcriptome-wide association analysis. These approaches can help in identifying causal variants and target genes at known AMD loci as well as could potentially reveal new candidates. SNP: Single nucleotide polymorphism; AMD: age-related macular degeneration; eQTL: expression quantitative trait loci.
DECLARATIONS
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipRatnapriya R is supported by Career Development Award from Research to Prevent Blindness (RPB) and Young Investigator Grant (M2021017N) from BrightFocus Foundation. This study is also supported by an Unrestricted grant from RPB to Baylor College of Medicine.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019;47:D1005-12.
2. Lichou F, Trynka G. Functional studies of GWAS variants are gaining momentum. Nat Commun 2020;11:6283.
3. Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet 2014;15:151-71.
4. Hellstrand K, Hermodsson S. An immunopharmacological analysis of adrenaline-induced suppression of human natural killer cell cytotoxicity. Int Arch Allergy Appl Immunol 1989;89:334-41.
5. Ratnapriya R, Swaroop A. Genetic architecture of retinal and macular degenerative diseases: the promise and challenges of next-generation sequencing. Genome Med 2013;5:84.
6. Friedman DS, O’Colmain BJ, Muñoz B, et al. Eye Diseases Prevalence Research Group. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 2004;122:564-72.
7. Klein R, Peto T, Bird A, Vannewkirk MR. The epidemiology of age-related macular degeneration. Am J Ophthalmol 2004;137:486-95.
8. Ferris FL, Davis MD, Clemons TE, et al. Age-Related Eye Disease Study (AREDS) Research Group. A simplified severity scale for age-related macular degeneration: AREDS Report No. 18. Arch Ophthalmol 2005;123:1570-4.
10. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 2001;119:1417-36.
11. Schultz DW, Klein ML, Humpert AJ, et al. Analysis of the ARMD1 locus: evidence that a mutation in HEMICENTIN-1 is associated with age-related macular degeneration in a large family. Hum Mol Genet 2003;12:3315-23.
12. Swaroop A, Chew EY, Rickman CB, Abecasis GR. Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu Rev Genomics Hum Genet 2009;10:19-43.
13. Weeks DE, Conley YP, Tsai HJ, et al. Age-related maculopathy: a genomewide scan with continued evidence of susceptibility loci within the 1q31, 10q26, and 17q25 regions. Am J Hum Genet 2004;75:174-89.
14. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005;308:385-9.
15. Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science 2005;308:421-4.
16. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005;308:419-21.
17. Priya RR, Chew EY, Swaroop A. Genetic studies of age-related macular degeneration: lessons, challenges, and opportunities for disease management. Ophthalmology 2012;119:2526-36.
18. Fritsche LG, Chen W, Schu M, et al. AMD Gene Consortium. Seven new loci associated with age-related macular degeneration. Nat Genet 2013;45:433-9, 439e1-2.
19. Fritsche LG, Igl W, Bailey JN, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 2016;48:134-43.
20. Singh N, Swaroop A, Ratnapriya R. Making biological sense of genetic studies of age-related macular degeneration. Adv Exp Med Biol 2021;1256:201-19.
21. Winkler TW, Grassmann F, Brandl C, et al. Genome-wide association meta-analysis for early age-related macular degeneration highlights novel loci and insights for advanced disease. BMC Med Genomics 2020;13:120.
22. Zwick ME, Cutler DJ, Chakravarti A. Patterns of genetic variation in Mendelian and complex traits. Annu Rev Genomics Hum Genet 2000;1:387-407.
23. Yang HJ, Ratnapriya R, Cogliati T, Kim JW, Swaroop A. Vision from next generation sequencing: multi-dimensional genome-wide analysis for producing gene regulatory networks underlying retinal development, aging and disease. Prog Retin Eye Res 2015;46:1-30.
24. Raychaudhuri S, Iartchouk O, Chin K, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet 2011;43:1232-6.
25. Helgason H, Sulem P, Duvvari MR, et al. A rare nonsynonymous sequence variant in C3 is associated with high risk of age-related macular degeneration. Nat Genet 2013;45:1371-4.
26. Seddon JM, Yu Y, Miller EC, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet 2013;45:1366-70.
27. Zhan X, Larson DE, Wang C, et al. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat Genet 2013;45:1375-9.
28. van de Ven JP, Nilsson SC, Tan PL, et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat Genet 2013;45:813-7.
29. Momozawa Y, Akiyama M, Kamatani Y, et al. Low-frequency coding variants in CETP and CFB are associated with susceptibility of exudative age-related macular degeneration in the Japanese population. Hum Mol Genet 2016;25:5027-34.
30. Corominas J, Colijn JM, Geerlings MJ, et al. Whole-exome sequencing in age-related macular degeneration identifies rare variants in COL8A1, a component of Bruch’s membrane. Ophthalmology 2018;125:1433-43.
31. Anderson DH, Radeke MJ, Gallo NB, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res 2010;29:95-112.
32. Stanton CM, Yates JR, den Hollander AI, et al. Complement factor D in age-related macular degeneration. Invest Ophthalmol Vis Sci 2011;52:8828-34.
33. Duvvari MR, Paun CC, Buitendijk GH, et al. Analysis of rare variants in the C3 gene in patients with age-related macular degeneration. PLoS One 2014;9:e94165.
34. Yu Y, Triebwasser MP, Wong EK, et al. Whole-exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum Mol Genet 2014;23:5283-93.
35. Saksens NT, Geerlings MJ, Bakker B, et al. Rare genetic variants associated with development of age-related macular degeneration. JAMA Ophthalmol 2016;134:287-93.
36. Pietraszkiewicz A, van Asten F, Kwong A, et al. Association of rare predicted loss-of-function variants in cellular pathways with sub-phenotypes in age-related macular degeneration. Ophthalmology 2018;125:398-406.
37. Huang LZ, Li YJ, Xie XF, et al. Whole-exome sequencing implicates UBE3D in age-related macular degeneration in East Asian populations. Nat Commun 2015;6:6687.
38. Geerlings MJ, Kremlitzka M, Bakker B, et al. The functional effect of rare variants in complement genes on C3b degradation in patients with age-related macular degeneration. JAMA Ophthalmol 2017;135:39-46.
39. Wagner EK, Raychaudhuri S, Villalonga MB, et al. Mapping rare, deleterious mutations in Factor H: association with early onset, drusen burden, and lower antigenic levels in familial AMD. Sci Rep 2016;6:31531.
40. Duvvari MR, van de Ven JP, Geerlings MJ, et al. Whole exome sequencing in patients with the cuticular drusen subtype of age-related macular degeneration. PLoS One 2016;11:e0152047.
41. Pras E, Kristal D, Shoshany N, et al. Rare genetic variants in Tunisian Jewish patients suffering from age-related macular degeneration. J Med Genet 2015;52:484-92.
42. Hoffman JD, Cooke Bailey JN, D’Aoust L, et al. Rare complement factor H variant associated with age-related macular degeneration in the Amish. Invest Ophthalmol Vis Sci 2014;55:4455-60.
43. Ratnapriya R, Acar İE, Geerlings MJ, et al. Family-based exome sequencing identifies rare coding variants in age-related macular degeneration. Hum Mol Genet 2020;29:2022-34.
44. Holz FG, Schmitz-Valckenberg S, Fleckenstein M. Recent developments in the treatment of age-related macular degeneration. J Clin Invest 2014;124:1430-8.
45. Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet 2015;47:856-60.
46. Pennesi ME, Neuringer M, Courtney RJ. Animal models of age related macular degeneration. Mol Aspects Med 2012;33:487-509.
47. Provis JM, Penfold PL, Cornish EE, Sandercoe TM, Madigan MC. Anatomy and development of the macula: specialisation and the vulnerability to macular degeneration. Clin Exp Optom 2005;88:269-81.
48. Adijanto J, Philp NJ. Cultured primary human fetal retinal pigment epithelium (hfRPE) as a model for evaluating RPE metabolism. Exp Eye Res 2014;126:77-84.
49. Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012;337:1190-5.
50. Deplancke B, Alpern D, Gardeux V. The genetics of transcription factor DNA binding variation. Cell 2016;166:538-54.
51. Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020;369:1318-30.
52. Strunnikova NV, Maminishkis A, Barb JJ, et al. Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum Mol Genet 2010;19:2468-86.
53. Pinelli M, Carissimo A, Cutillo L, et al. An atlas of gene expression and gene co-regulation in the human retina. Nucleic Acids Res 2016;44:5773-84.
54. Hoshino A, Ratnapriya R, Brooks MJ, et al. Molecular anatomy of the developing human retina. Dev Cell 2017;43:763-79.e4.
55. Yan W, Peng YR, van Zyl T, et al. Cell atlas of the human fovea and peripheral retina. Sci Rep 2020;10:9802.
56. Sridhar A, Hoshino A, Finkbeiner CR, et al. Single-cell transcriptomic comparison of human fetal retina, hPSC-derived retinal organoids, and long-term retinal cultures. Cell Rep 2020;30:1644-59.e4.
57. Cowan CS, Renner M, De Gennaro M, et al. Cell types of the human retina and its organoids at single-cell resolution. Cell 2020;182:1623-40.e34.
58. Voigt AP, Mulfaul K, Mullin NK, et al. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc Natl Acad Sci U S A 2019;116:24100-7.
59. Liang Q, Dharmat R, Owen L, et al. Single-nuclei RNA-seq on human retinal tissue provides improved transcriptome profiling. Nat Commun 2019;10:5743.
60. Gamazon ER, Segrè AV, van de Bunt M, et al. GTEx Consortium. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 2018;50:956-67.
61. Ongen H, Brown AA, Delaneau O, Panousis NI, Nica AC, Dermitzakis ET. GTEx Consortium. Estimating the causal tissues for complex traits and diseases. Nat Genet 2017;49:1676-83.
62. Kim-Hellmuth S, Aguet F, Oliva M, et al. GTEx Consortium. Cell type-specific genetic regulation of gene expression across human tissues. Science 2020;369:eaaz8528.
63. Raj T, Rothamel K, Mostafavi S, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 2014;344:519-23.
64. Watanabe K, Umićević Mirkov M, de Leeuw CA, van den Heuvel MP, Posthuma D. Genetic mapping of cell type specificity for complex traits. Nat Commun 2019;10:3222.
65. Wang X, Park J, Susztak K, Zhang NR, Li M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun 2019;10:380.
66. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220.
67. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453-7.
68. Fuzik J, Zeisel A, Máté Z, et al. Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat Biotechnol 2016;34:175-83.
69. Grün D, Lyubimova A, Kester L, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015;525:251-5.
70. Jaitin DA, Kenigsberg E, Keren-Shaul H, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014;343:776-9.
71. Regev A, Teichmann SA, Lander ES, et al. Human Cell Atlas Meeting Participants. The human cell atlas. Elife 2017;6:e27041.
72. Mathys H, Davila-Velderrain J, Peng Z, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019;570:332-7.
73. Agarwal D, Sandor C, Volpato V, et al. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat Commun 2020;11:4183.
74. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008;5:621-8.
75. Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet 2019;20:631-56.
76. Wang Y, Navin NE. Advances and applications of single-cell sequencing technologies. Mol Cell 2015;58:598-609.
77. Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet 2018;19:491-504.
78. Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 2010;6:e1000888.
79. Nica AC, Montgomery SB, Dimas AS, et al. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet 2010;6:e1000895.
80. Ratnapriya R, Sosina OA, Starostik MR, et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat Genet 2019;51:606-10.
81. Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet 2015;16:197-212.
82. Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet 2009;10:184-94.
83. Gilad Y, Rifkin SA, Pritchard JK. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet 2008;24:408-15.
84. Hukku A, Pividori M, Luca F, Pique-Regi R, Im HK, Wen X. Probabilistic colocalization of genetic variants from complex and molecular traits: promise and limitations. Am J Hum Genet 2021;108:25-35.
85. Strunz T, Kiel C, Grassmann F, et al. A mega-analysis of expression quantitative trait loci in retinal tissue. PLoS Genet 2020;16:e1008934.
86. Orozco LD, Chen HH, Cox C, et al. Integration of eQTL and a single-cell atlas in the human eye identifies causal genes for age-related macular degeneration. Cell Rep 2020;30:1246-59.e6.
87. Liu B, Calton MA, Abell NS, et al. Genetic analyses of human fetal retinal pigment epithelium gene expression suggest ocular disease mechanisms. Commun Biol 2019;2:186.
88. White MJ, Yaspan BL, Veatch OJ, Goddard P, Risse-Adams OS, Contreras MG. Strategies for pathway analysis using GWAS and WGS data. Curr Protoc Hum Genet 2019;100:e79.
89. Waksmunski AR, Grunin M, Kinzy TG, Igo RP Jr, Haines JL, Cooke Bailey JN. International Age-Related Macular Degeneration Genomics Consortium. Pathway analysis integrating genome-wide and functional data identifies PLCG2 as a candidate gene for age-related macular degeneration. Invest Ophthalmol Vis Sci 2019;60:4041-51.
90. Sekar S, McDonald J, Cuyugan L, et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol Aging 2015;36:583-91.
91. Fromer M, Roussos P, Sieberts SK, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 2016;19:1442-53.
92. Tian L, Kazmierkiewicz KL, Bowman AS, Li M, Curcio CA, Stambolian DE. Transcriptome of the human retina, retinal pigmented epithelium and choroid. Genomics 2015;105:253-64.
93. Newman AM, Gallo NB, Hancox LS, et al. Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med 2012;4:16.
94. Pauly D, Agarwal D, Dana N, et al. Cell-type-specific complement expression in the healthy and diseased retina. Cell Rep 2019;29:2835-48.e4.
95. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 2005;4:Article17.
96. Calabrese GM, Mesner LD, Stains JP, et al. Integrating GWAS and co-expression network data identifies bone mineral density genes SPTBN1 and MARK3 and an osteoblast functional module. Cell Syst 2017;4:46-59.e4.
97. Gustafsson M, Gawel DR, Alfredsson L, et al. A validated gene regulatory network and GWAS identifies early regulators of T cell-associated diseases. Sci Transl Med 2015;7:313ra178.
98. Mäkinen VP, Civelek M, Meng Q, et al. Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis (CARDIoGRAM) Consortium. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet 2014;10:e1004502.
99. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559.
100. Gusev A, Ko A, Shi H, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 2016;48:245-52.
101. Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Statist Med 2008;27:1133-63.
102. Peng YR, Shekhar K, Yan W, et al. Molecular classification and comparative taxonomics of foveal and peripheral cells in primate retina. Cell 2019;176:1222-37.e22.
103. Menon M, Mohammadi S, Davila-Velderrain J, et al. Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat Commun 2019;10:4902.
104. van der Wijst M, de Vries DH, Groot HE, et al. The single-cell eQTLGen consortium. Elife 2020;9:e52155.
105. Sisternes L, Simon N, Tibshirani R, Leng T, Rubin DL. Quantitative SD-OCT imaging biomarkers as indicators of age-related macular degeneration progression. Invest Ophthalmol Vis Sci 2014;55:7093.
106. Niu S, de Sisternes L, Chen Q, Rubin DL, Leng T. Fully automated prediction of geographic atrophy growth using quantitative spectral-domain optical coherence tomography biomarkers. Ophthalmology 2016;123:1737-50.
107. Lai TT, Hsieh YT, Yang CM, Ho TC, Yang CH. Biomarkers of optical coherence tomography in evaluating the treatment outcomes of neovascular age-related macular degeneration: a real-world study. Sci Rep 2019;9:529.
108. Lambert NG, ElShelmani H, Singh MK, et al. Risk factors and biomarkers of age-related macular degeneration. Prog Retin Eye Res 2016;54:64-102.
109. Lauwen S, de Jong EK, Lefeber DJ, den Hollander Al. Omics biomarkers in ophthalmology. Invest Ophthalmol Vis Sci 2017;58:BIO88-98.
110. Kersten E, Paun CC, Schellevis RL, et al. Systemic and ocular fluid compounds as potential biomarkers in age-related macular degeneration. Surv Ophthalmol 2018;63:9-39.
111. Bailey JN, Hoffman JD, Sardell RJ, Scott WK, Pericak-Vance MA, Haines JL. The application of genetic risk scores in age-related macular degeneration: a review. J Clin Med 2016;5:31.
112. Heesterbeek TJ, Lorés-Motta L, Hoyng CB, Lechanteur YTE, den Hollander AI. Risk factors for progression of age-related macular degeneration. Ophthalmic Physiol Opt 2020;40:140-70.
113. Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med 2020;12:44.
114. Ding Y, Liu Y, Yan Q, et al. AREDS2 Research Group. Bivariate analysis of age-related macular degeneration progression using genetic risk scores. Genetics 2017;206:119-33.
115. Peng Y, Keenan TD, Chen Q, et al. Predicting risk of late age-related macular degeneration using deep learning. NPJ Digit Med 2020;3:111.
116. Yan Q, Weeks DE, Xin H, et al. Deep-learning-based prediction of late age-related macular degeneration progression. Nat Mach Intell 2020;2:141-50.
117. Geerlings MJ, de Jong EK, den Hollander AI. The complement system in age-related macular degeneration: a review of rare genetic variants and implications for personalized treatment. Mol Immunol 2017;84:65-76.
118. Ratnapriya R. Applications of genomic technologies in retinal degenerative diseases. Adv Exp Med Biol 2019;1185:281-5.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ratnapriya R. Transcriptomics insights into interpreting AMD-GWAS discoveries for biological and clinical applications. J Transl Genet Genom 2022;6:240-56. http://dx.doi.org/10.20517/jtgg.2021.54
AMA Style
Ratnapriya R. Transcriptomics insights into interpreting AMD-GWAS discoveries for biological and clinical applications. Journal of Translational Genetics and Genomics. 2022; 6(2): 240-56. http://dx.doi.org/10.20517/jtgg.2021.54
Chicago/Turabian Style
Ratnapriya, Rinki. 2022. "Transcriptomics insights into interpreting AMD-GWAS discoveries for biological and clinical applications" Journal of Translational Genetics and Genomics. 6, no.2: 240-56. http://dx.doi.org/10.20517/jtgg.2021.54
ACS Style
Ratnapriya, R. Transcriptomics insights into interpreting AMD-GWAS discoveries for biological and clinical applications. J. Transl. Genet. Genom. 2022, 6, 240-56. http://dx.doi.org/10.20517/jtgg.2021.54
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.