
Genetic approaches and pathogenic pathways in the clinical management of Charcot-Marie-Tooth disease


Abstract
Charcot-Marie-Tooth (CMT) disease is the most common inherited neuromuscular disorder, affecting at least 1 in 2500 individuals. CMT refers to a heterogeneous group of inherited neuropathies from both phenotypic and genetic points of view. Over the last decades, there have been important advances not only in the identification of causative genes but also in understanding the molecular basis for many forms of CMT. In fact, to date, around 100 genes have been related to CMT disease, thanks to next generation sequencing techniques, and they have been proven to affect either the myelin or axon of peripheral nerves. Moreover, its genetic diagnosis has remarkedly improved, although there are still difficulties when it comes to treatment. In this review, we explore in depth the eight most prevalent genes associated with CMT: GDAP1, GJB1, HINT1, MFN2, MPZ, PMP22, SH3TC2, and SORD. We also address the disrupted cellular processes and pathophysiological mechanisms involved in the disease. A better understanding of the pathogenic mechanisms responsible for each type of CMT would be essential to identifying molecular targets and therapeutic strategies.
Keywords
THE CLINICAL AND GENETIC FEATURES
Charcot-Marie-Tooth (CMT) disease was described and named in 1886 by Charcot, Marie, and Tooth[1,2]. Originally described as peroneal muscular atrophy, CMT is a hereditary motor and sensory neuropathy (HMSN) that primarily affects either myelin or the axon of peripheral nerves[3]. This review is focused on CMT as well as other related genetic neuropathies, which include distal hereditary motor neuropathies (dHMN) with minimal or absent sensory involvement, and hereditary sensory and autonomic neuropathies (HSN and HSAN), with significant sensory involvement.
CMT is the most common inherited disorder of the peripheral nervous system (PNS), with an estimated prevalence of 28-40 individuals per 100,000 inhabitants, with global distribution and no ethnic predisposition[4-6]. Individuals with CMT show symmetric, slowly progressive in a length-dependent manner, distal neuropathy of the legs and arms. CMT disease usually begins in the first to third decade of life, causing slowly progressive distal muscle weakness and atrophy, weak ankle dorsiflexion, depressed tendon reflexes, and pes cavus deformity[7-11]. Other symptoms are ataxia, pyramidal signs, and hypoacusia. More aggressive phenotypes, in which the symptoms appear within the first two years of life, are characterized by hypotonia, areflexia, and ataxia that represent greater disability in patients, and in some cases, limit the patient’s autonomous ambulation and can lead to significant motor disability[12].
CMT types and classification
There is a remarkable heterogeneity in the spectrum of CMT and related disorders. In the very first instance, heterogeneity can be observed in symptomatology and severity of the disease[3,8]. Once the symptoms are detected, ancillary tests (electrophysiologic and neuropathologic studies) help clinicians to classify patients into those with demyelinating or axonal primary involvement and, in some cases, intermediate forms[13]. Clinical or phenotypic heterogeneity is complemented with high genetic heterogeneity[3,14]. Since the discovery of the 1.4 Mb duplication in chromosome 17p11.2[15,16], the number of CMT-associated genes has been increasing, and today almost 100 genes causing CMT disease and other genetic neuropathies are now known. The identification of the responsible gene and its inheritance pattern also helps the clinical practice make a correct classification of patients[7,17]. The observed inheritance patterns include autosomal dominant, autosomal recessive, and X-linked (partially dominant and recessive) forms. However, many patients present apparent sporadic diseases, attributable to de novo mutations[18].
We can distinguish two CMT neuropathies according to the type of cells primarily affected and the nerve conduction velocities (NCVs): demyelinating CMT which affects the myelin-forming Schwann cells and with NCVs below 38 m/s, and axonal CMT which affects the axons of neurons and usually presents NCVs above 38 m/s[13,19]. Intermediate forms with overlapping demyelinating and axonal features, especially within the same family, are defined by NCVs lying between 25 and 45 m/s[3,4,7,13]. Taking together the conduction velocity parameters and the mode of inheritance, we can stratify CMT into five different categories: demyelinating plus autosomal dominant inheritance (CMT1); axonal plus autosomal dominant or recessive (CMT2); demyelinating plus autosomal recessive (CMT4); and X-linked (dominant or recessive) (CMTX). The term CMT3 has been reserved to designate Dejerine-Sottas syndrome or neuropathy, which is a specific category related to a congenital or infantile-onset and a severe (usually demyelinating) phenotype. Further subdivision of these CMT types is based mainly on causative genes and assigned loci.
The increasing knowledge of CMT genetics and pathophysiology has led to changes in the way the different types were classified initially. Some efforts have been made to simplify its nomenclature, reducing the risk of denomination errors[20,21], since the discovery of newly associated genes susceptible to being the cause made the classification more and more difficult. Specifically, Mathis et al. in 2015, opened a way for a precise denomination including: (1) the inheritance pattern; (2) the pathophysiological phenotype (by using “de” or “ax” for demyelinating and axonal forms, respectively); and (3) giving more importance to the causal gene[21]. Based on it, CMT1A 17p11.2 duplication would be AD-CMTde-PMP22dup or CMT4A would become AR-CMTde-GDAP1[21]. This is an informative denomination that, in fact, is helpful and understandable for both patients and clinicians. These types of proposals are an example of all the efforts that the scientific community has made to manage such a heterogeneous group of diseases[20]. In any case, in our ambit, we still maintain the traditional nomenclature that, moreover, can be found in clinical databases such as OMIM. This is the reason this denomination is used in this review.
The molecular genetics of CMT began in 1991 with the discovery of the 1.4 Mb duplication in the short arm of chromosome 17, which contains the dose-sensitive peripheral myelin protein 22 (PMP22) gene, causing CMT1A and became the most common cause of genetic neuropathies[15,22]. By 1992, point or indel mutations in GJB1 (encoding connexin 32 [Cx32]), PMP22, and MPZ had also been discovered[17,23]. Loci and genes for CMT and related peripheral neuropathies were initially identified using linkage studies, positional cloning, or candidate gene approaches[22]. Since the publication of the first draft of the human genome in 2001, the development of high-throughput technologies, such as genome mapping, whole-exome sequencing, and whole-genome sequencing, have accelerated the gene and mutation discovery in CMT research[14,23]. As the Human Genome Project reached completion, the identification of CMT genes increased markedly, so the number of genes associated with the disease has been increasing in the last years.
Currently, around 100 genes have been identified in Mendelian inheritance of genetic neuropathies. Although there are myriad gene associations and pathophysiologic mechanisms, it is clear that the mutations associated with the disease are closely related to the formation, compaction, and maintenance of myelin (PMP22, P0, Cx32, EGR2, NDGR1, PRX, etc.), the neuronal soma, axon and cytoskeleton conservation (NEFL, LMNA, MORC2, etc.), the axonal transport (RAB7), and the mitochondrial dynamics (MFN2, GDAP1, GARS, HSP22, HSP27, etc.)[3,7]. This means that, independently of the defect (metabolic, cytoplasmic, or structural) that primarily affects the myelin or axon, as well as the Schwann cell-axon structure, the axonal degenerative process is the final common pathway in neuropathies that primarily affect the largest and longest fibers[24].
CMT genes and their corresponding proteins have been classified according to their localization in the neuron, and general information about their main biological function and proposed pathomechanisms is explained here. The well-established proteins and their proposed pathogenic mechanisms for the nerve (either in myelin or in the neuronal axon and soma) are summarized in Supplementary Table 1. The code of the MIM genes (*) and their mode of inheritance are indicated together with the MIM phenotype (#) of the neuropathy caused by the pathogenic variants of the corresponding gene. Information is updated each year in the context of all neuromuscular disorders[25].
Genetic diagnosis
Due to the high heterogeneity in terms of gene associations, the approach to the diagnosis of CMT and genetic neuropathies has evolved from a purely clinical approach in the past to a combined clinical/genetic approach. As happens in many other diseases, genetic testing can also be used on the patient and their family for predictive, antenatal, or preimplantation assessment.
Because CMT1A (PMP22) duplication represents 50% of CMT patients (around 70.7% of demyelinating CMT), the very first step is to analyze them by multiplex ligation-dependent probe amplification (MLPA). This is the current technique for genetic testing of the CMT1A duplication (and hereditary neuropathy with liability to pressure palsies or HNPP deletion) in postnatal and prenatal diagnosis. However, in the case of preimplantation genetic diagnosis (PGD), segregation analysis of CMT1A linked microsatellites is still a molecular technique for genetic testing. After that, if negative, different algorithms help to perform sequential testing of individual genes using Sanger sequencing[26,27]. In this scenario, the most promising candidate gene is analyzed and, if negative, the next most likely candidate is tested. Several publications provide the scientific community with algorithms and pipelines designed to maximize efficiency when diagnosing CMT disease[14,27].
Nevertheless, this is a very expensive and time-consuming workflow, especially in those cases where the causative gene is individually rare. With the evolution of next generation sequencing (NGS, or massive parallel sequencing) techniques, it is now possible to analyze all CMT genes by a selection of genes (panels), the exome (containing only the protein-coding sequences), or the genome, and this strategy has become the most cost-efficient approach[14,28,29]. This means that, as technology has advanced and the cost has dropped, these approaches have replaced the traditional screening gene by gene. Therefore, NGS technologies have helped not only to identify genes in association with CMT but also to develop more efficient workflows to couple a clinical diagnosis with the genetic diagnosis.
However, some challenges remain to be solved and are responsible for deficits in the diagnostic rates. A critical issue in this two-step process (clinical exam and genetic testing) is the analysis and interpretation of genomic data generated by NGS[29]. Depending on different criteria, geneticists have to evaluate the relationship between a variant and the described phenotype to identify causative variants. Sometimes, the chosen criteria allow them to find a variant that could be considered causative in achieving the etiological diagnosis. However, the same workflow may be inconclusive, generally because of the identification of variants of uncertain clinical significance or VUS [according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP)][30].
Treatment
Historically, inherited peripheral neuropathies have been challenging to treat. There is little specific therapy for these neuropathies other than genetic counseling as well as symptomatic treatment, physical therapy, and rehabilitation[31,32]. This represents supportive treatment, limited to rehabilitative therapy and surgical treatment of skeletal deformities and/or abnormalities of soft tissues[33]. There are no established disease-modifying therapies to date. In clinical practice, patients are often evaluated and managed by a multidisciplinary team that includes adult and pediatric neurologists, physiatrists, orthopedic surgeons, and physical and occupational therapists[34].
On the one hand, these supportive treatments are usually based on rehabilitation to improve patient’s posture and balance and shoe modifications, orthoses, or other assistive devices that have been shown to improve patient’s walking economy[35]. Although data are limited, there is evidence that mild to moderate exercise is effective and safe for CMT patients and that it could be interesting to consider it as a therapeutic intervention[36]. Moreover, many different approaches have been used to treat skeletal deformities (especially of the feet) from a surgical point of view[37].
On the other hand, different pharmacological therapies have been considered in the treatment of CMT patients. First, a symptomatic drug therapy approach can be selected: pain can be an emerging feature of CMT (usually an osteo-arthropathic type of pain). This treatment includes physical therapy, as explained above, but also drugs (both for neuropathic and non-neuropathic pain)[35,38,39]. Then, specific drug therapies have been considered in the CMT research, mostly those related to commonly found mutated genes[40-43], and several experimental models have been useful to explore different approaches (Available from: https://www.jax.org/search?q=Charcot%20Marie%20Tooth).
Some clinical trials with pharmacologic agents aimed at reducing PMP22 expression in CMT1A patients, such as ascorbic acid, which did not improve the patient condition[44]. After preclinical assays in a CMT1A rat model, the progesterone receptor antagonist onapristone was discarded because it was not safe in humans[45]. Recently, high-dose PXT3003 (baclofen/naltrexone/D-sorbitol) has demonstrated significant therapeutic effects in patients with CMT1A and has emerged as a promising treatment option[46]. HDAC6 inhibitors have shown positive effects on axonal defects in mouse models of several forms of CMT[47-49], opening a new window in the pharmacological treatment of CMT disease[50,51]. Antioxidant therapy with either the veterinary antibiotic florfenicol or mitoQ has also shown effectiveness in a knockout model of GDAP1-related CMT when starting administration very early in mouse life, but not in older mice[52]. Gene therapy research in animal models is becoming more relevant. AAV9-mediated Schwann cell-targeted gene therapy of Gjb1-null mouse improves motor performance and sciatic nerve conduction velocities along with improved myelination and inflammatory processes in peripheral nerve tissues[53]. Therefore, much work remains to be done for the treatment of CMT. The discovery of cellular mechanisms underlying the disease pathophysiology opens new options for preclinical studies searching for new treatments that include drug repositioning[52]. An in-depth overview of current research in the pharmacological and biological treatment of CMT neuropathies is beyond the scope of this review. The current status of therapeutic investigations and ongoing clinical trials in CMT disease and genetic neuropathies was recently reported by
CMT therapeutics: target cell processes and pathways and drug strategies
| Cell process or mechanism of action | Compound/therapeutic strategy |
| Reduction of gene expression | Ascorbic acid |
| Reduction of protein synthesis | Progesterone antagonists and modulators |
| Inhibition of Schwann cells proliferation and reduction of protein synthesis (baclofen: GABAB receptor modulator) | PXT3003 (baclofen, sorbitol, naltrexone) |
| Partial silencing of gene expression | Gene silencing |
| Gene insertion (AAV1-NT3) | Gene therapy |
| Gene substitution | |
| Regulation of myelin thickness | Neuregulin pathways |
| UPR inhibition | Curcumin, sephin-1 |
| TRPA1 and TRPV1 channels activation | FLX-787 |
| Prevention of axonal degeneration | SARM1 inhibitors |
| Reduction of microtubules acetylation (axonal transport) | HDAC6 inhibitors |
| Myostatin pathway | ACE-083 |
| Reduction of abnormal calcium influx in Schwann cells | P2X7 receptor modulators |
| Correction of defective lipid biosynthesis | Dietary lipid supplementation |
| Nav 1.8 channel blocking | Sodium channel blockers |
| Decreased number/activity of macrophages in the nerve | CSF1R inhibitors |
| PIKfyve inhibition and decrease of PI3,5P2 levels | PIKfyve enzyme inhibitors |
| Reduction of neurotoxic deoxysphingolipids | L-Serine |
| Purine nucleotides supply | S-adenosylmethionine (SAM) |
| Inhibition of aldose reductase | Aldose reductase inhibitors |
PATHOGENIC MECHANISMS AND NERVE PATHOPHYSIOLOGY
CMT and related diseases constitute a very heterogeneous group of disorders. From clinical to genetic diagnosis, variability among patients is very high and pathogenic mechanisms involve Schwann cell and neuron/axon pathways. Figure 1 and Supplementary Table 1 provide information on CMT genes and illustrate the cellular and molecular mechanisms that compromise the nerve physiology, either in the myelin sheath or the neuron soma and axon.
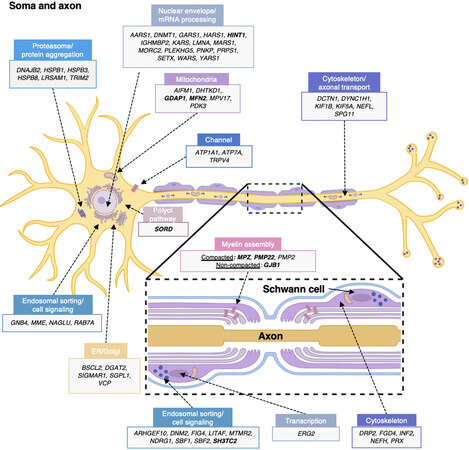
Figure 1. Schematic summary of genes causing CMT hereditary neuropathy. Gene products are assigned either to the neuron body and axon or to the myelinating Schwann cell (cross-sectional view in box). Proteins are classified according to their main functions or pathways and their proposed pathomechanisms in CMT. The most commonly involved genes are indicated in bold. More information about each gene and/or protein is available in Table 2 and Supplementary Table 1. ER: Endoplasmic reticulum. Illustration created with BioRender (Available from: https://biorender.com/).
The following sections explain, in more detail and alphabetical order, the pathogenic mechanisms related to the genes that most frequently constitute the genetic cause of CMT. Together with this detailed description, Table 2 summarizes this information and correlates with Figure 2, which illustrates in detail the subcellular localization of the proteins and the mechanisms and pathways affected by the pathogenic variants.
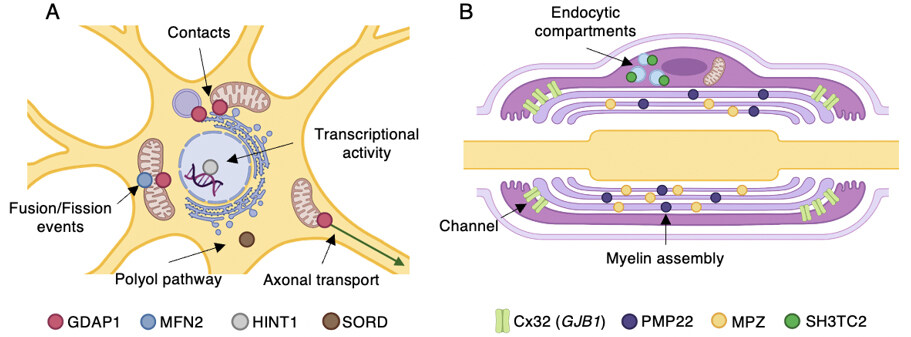
Figure 2. Most common CMT-associated genes and their proposed pathomechanisms. Subcellular localization and the main functions of gene products are indicated. (A) Diagram showing a neuron soma. (B) A myelinating Schwann cell. Illustration created with BioRender (Available from: https://biorender.com/).
Most common CMT genes: inheritance, phenotype, and cell pathophysiology
| Gene | OMIM (*) | MoI | Phenotype neuropathy type | OMIM (#) | Localization | Protein function/pathway | Disrupted process | ||||
| Schwann cell | Soma/axon | ||||||||||
| A | D | I | |||||||||
| Most commonly involved genes | |||||||||||
| GDAP1 | 606598 | AR AD | √ | √ | √ | AR-CMT2K CMT2K CMT4A | 607706 607831 214400 | √ | Mitochondria fission | Mitochondria | |
| GJB1 | 304040 | XL | √ | √ | √ | CMTX | 302800 | √ | Cx32: gap junction formation + myelin assembly and transport | Channel | |
| HINT1 | 601314 | AR | √ | Neuromyotonia Axonal neuropathy | 137200 | √ | Modulation of transcriptional activity | Nuclear envelope, mRNA processing | |||
| MFN2 | 608507 | AR AD | √ | CMT2A2A CMT2A2B CMT6A | 609260 617087 601152 | √ | Mitochondrial fusion | Mitochondria | |||
| MPZ | 159440 | AD | √ | √ | √ | CMT1B DI-CMTD CMT2I CMT2J | 118200 607791 607677 607736 | √ | Myelin assembly | Myelin assembly | |
| PMP22 | 601097 | AD | √ | CMT1A CMT1E | 118220 118300 | √ | Myelin assembly | Myelin assembly | |||
| SH3TC2 | 608206 | AR | √ | CMT4C | 601596 | √ | Targets to intracellular endosome recycling | Endosomal sorting and cell signaling | |||
| SORD | 182500 | AR | √ | √ | Peripheral neuropathy | 618912 | √ | Polyol pathway | Polyol pathway (cytosolic) | ||
GDAP1
Among the most commonly involved genes in the pathogenesis of CMT, we can find GDAP1. This gene encodes ganglioside-induced differentiation-associated protein 1 (GDAP1), which is an atypical glutathione S-transferase (GST)[54] with glutathione-conjugating and membrane-remodeling functions[55]. This protein can be found in the outer mitochondrial membrane (OMM) and the mitochondria-associated membranes (MAMs), and it is mainly expressed in neurons[56-58].
The scenario of GDAP1 mutations is quite different from other CMT disease-causing genes due to its heterogeneity: mutations in this gene have been related to axonal forms (AR-CMT2K with vocal cord paresis (CMT2K), an intermediate form (CMTRIA), and a demyelinating (CMT4A) form of the disease. Both recessive and dominant modes of inheritance have been reported[56]. To date, more than 100 mutations in GDAP1 gene have been related to CMT phenotype[59].
Clinically, CMT caused by mutations in this gene is characterized by severe distal motor and sensory neuropathy. Nevertheless, a diverse spectrum of phenotypes should be considered due to the variability in inheritance patterns[56,60,61]. It has been proposed that, while autosomal recessive mutations are responsible for early-onset and severe neuropathies, dominant mutations cause a milder course of the disease[62]. Regarding the histological abnormalities, the sural nerve biopsy of severe affected segregating the disease in an autosomal recessive manner shows a pronounced depletion of myelinated fibers, regenerative clusters and signs of axonal atrophy. Additionally, a small proportion of thin myelinated fibers and proliferation of Schwann cells forming onion bulb structures have also been found. The most relevant cytoplasmic feature would be the mitochondrial abnormalities[63].
Although the explicit molecular mechanism underlying the GDAP1 function remains unclear, several studies have explored its role in mitochondria physiology: morphology, function, and dynamics[59]. Firstly, the consequence of GDAP1 mutations can impair these mitochondrial functions through mitochondrial membrane potential reduction, ATP production changes, or a disbalance of their dynamics[57,64,65]. Second, GDAP1 may also interact with transport proteins involved in mitochondrial transport and movement. Therefore, the alteration of this process could be an explanation for the axonal loss that can be seen in CMT patients carrying GDAP1 mutations[56]. Finally, the recently established relationship between GDAP1 and mitochondrial-associated membranes (MAMs) would support the idea that GDAP1 mutations could affect the formation and functioning of the ER-mitochondria contacts[66]. This would explain the alteration of store-operated Ca2+ entry (SOCE) and calcium homeostasis, together with mitochondrial dynamics and transport, too. On the other hand, recent studies have shown that GDAP1 participates in membrane contact sites (MCSs) between the mitochondria and the lysosome, supporting the idea that GDAP1 enables the proper function of mitochondrial MCSs[62,67]. Finally, it has been reported that it also influences the structure and probably the function of the Golgi apparatus[59]. In addition, functional studies have characterized the phenotype derived from GDAP1 mutations. As expected, these include the disruption of mitochondrial fission-fusion events, changes in mitochondrial distribution, impairment of the mitochondrial membrane potential, increases in the concentration of reactive oxygen species, reductions in glutathione content, and alteration in the bioenergetics of mitochondria[57,64,65,68-71].
GJB1
The X-linked form of Charcot-Marie-Tooth disease (CMT1X) is the second most common form of HMSN[13,72,73] and accounts for 90% of all CMTX cases[74]. This form of the disease is caused by mutations in GJB1 gene, which encodes the gap junction protein connexin32 (Cx32). GJB1 disorders are typically characterized by peripheral motor and sensory neuropathy with or without fixed central nervous system abnormalities and/or acute, self-limited episodes of transient neurologic dysfunction[75]. Peripheral neuropathy typically manifests in affected males between ages 5 and 25 years. Although both men and women are affected, males have moderate to severe symptoms, while heterozygous females may remain asymptomatic or may have mild CMTX1, being the skewed X-inactivation the likely explanation for the reduced severity. It should be noted that in those cases where women are severely affected, the most promising explanation is the non-random X-inactivation in each myelinating cell[76].
It should be noted that the nerve conduction velocities in patients with CMT-GJB1 are in the intermediate range: faster than in patients with the demyelinating forms (CMT1) and slower than axonal/neuronal forms (CMT2); however, in female cases, NCVs are often preserved. Moreover, median nerve conductions appear to be generally more severely affected than those of the ulnar nerve[77,78]. Finally, in nerve biopsies, the most prominent finding is the increasing number of regenerated axon clusters, together with an age-related loss of myelinated fibers[79].
The GJB1 gene encodes a member of a large family of connexins. Specifically, GJB1 encodes connexin 32, a protein that is remarkedly present in Schwann cells and oligodendrocytes, which are the myelinating glia of the peripheral and central nervous systems, respectively[74]. This explains why patients with CMT1X can have both central and PNS manifestations.
Six connexins molecules form a hemichannel, arranged around a central pore. Two of these hemichannels form a gap junction channel, allowing the formation of a link between two cells. Tens to thousands of these connexin channels are localized along the cell membrane, creating what is known as gap junction plaques[72]. Thanks to these channels, ions, signaling molecules, and/or small metabolites can diffuse between cells, helping them to couple in electrical and chemical ways, between other functions[74]. In Schwann cells, Cx32 is localized to the non-compact myelin of the paranodes and Schmidt-Lantermann incisures, where it forms gap junctions between adjacent loops of non-compact myelin, predicted to provide a radial diffusion pathway[80-85]. This means that, in Schwann cells, Cx32 forms a reflexive pathway between the abaxonally located nucleus of the Schwann cells and the adaxonal region[80].
Since the first report that mutations in GJB1 cause CMT1X, more than 400 mutations were predicted to impact protein function. Many of them could be predicted to cause loss-of-function effect, although the mechanisms could differ: nonsense or frameshift mutations that affect the N-terminal part, mutations that affect the promoter region[86,87], or large deletions involving the entire coding region of the gene would not be expected to produce functional channels[88].
The biological mechanisms by which the different mutations can lead to this total or partial loss of function are diverse: reduction of the minimal luminal dimension (that would affect the diffusion of specific small molecules)[89], increasing sensitivity to acidification-induced closure, or stabilization of the closed state of the channel[90] are some examples of the biophysical alterations that have been explored. Moreover, it has been analyzed how the junctional coupling can be affected by mutations in this gene, due to reduced steady-state levels of the protein[90].
Although the most common pathogenic mechanism of GJB1 mutations is this loss-of-function, some studies have reported gain-of-function mechanisms of some Cx32 mutants[91]. Overall, clinical studies suggest that the peripheral manifestations of CMT1X are likely to be due to loss-of-function, while in the central nervous system gain-of-function may contribute[80].
HINT1
In 2012, the histidine triad nucleotide-binding protein 1 gene (HINT1) was identified in recessive forms of axonal CMT (accounting for 10% of recessive CMT patients). At that time, it was reported that around 80% of patients carrying HINT1-causative variants showed neuromyotonia[92]. This phenotypic sign guided the clinical diagnosis of these cases, and it was considered that HINT1-associated peripheral neuropathy represented a distinct clinical and genetic entity that needed to be differentiated from other CMT types[93] described as axonal, motor-greater-than-sensory polyneuropathy with a childhood-onset, combined with neuromyotonia[92,94-96]. However, the identification of more patients and/or causative mutations extended the clinical spectrum. From the histological point of view, sural nerve biopsies of some reported cases showed axonal neuropathy, even in the absence of clinical sensory abnormalities.
Interestingly, HINT1 has a non-random geographic distribution of patients. The majority of causative mutations are found in Central and Eastern European individuals[97], and this has been attributed to three different founder mutations, p.Arg37Pro, p.Cys84Arg, and p.His112Asn.
HINT1 encodes a member of the histidine triad (HIT) protein family. Specifically, it is a globular protein that acts as a homodimer and binds purine nucleosides and nucleotides. Both its HIT motif and C-terminal loop are essential to establishing nucleotide contacts and maintaining substrate specificity[98], respectively. Furthermore, dimerization is required to maintain catalytic activity[98]. Several studies have reported that HINT1 mutations cause a loss-of-function effect through several mechanisms. For instance, they can affect critical residues for catalytic activity, they can be related to nonsense mediated decay of mRNA, or they can cause protein instability and, consequently, proteasome-mediated degradation[92].
MFN2
CMT type 2A is the most common axonal form of CMT and is caused almost exclusively by mutations in the MFN2 gene[99]. This type of CMT is characterized by peripheral neuropathy that can also involve the central nervous system[100]. Mutations in MFN2 usually have an autosomal dominant pattern of inheritance, but, occasionally, MFN2 mutations can be recessive or even semidominant.
About 100 mutations in MFN2 related to CMT2A2 have been described[101], but the exact relationship between the genotype and the phenotype of MFN2 patients remains to be clarified. Interestingly, a childhood onset of autosomal dominant CMT2A2 is the most predictive marker of significant disease severity[102]. In general, in comparison to demyelinating CMT1A, axonal CMT2A2 is more severe and shows a motor-predominant phenotype that usually carries a greater burden of disability[103,104]. MFN2 neuropathy can also be associated with sensorineural hearing loss, optic atrophy, and, in some cases, cerebellar atrophy, spastic paraparesis, and cognitive deficits.
Mitofusin-2 and -1 are homologous proteins, members of a mitochondrial transmembrane GTPase family. They show ubiquitous expression in eukaryotic cells, where they play a role in the dynamic mitochondrial remodeling process[102]. Both of them are known to play a critical role in the mitigation of mitochondrial stress, helping to maintain mitochondrial “quality control” and facilitate apoptosis if necessary (under severe cellular stress)[105].
Mitofusin-2 has two main functions: the promotion of mitochondrial fusion and the mediation of endoplasmatic reticulum (ER)-mitochondrial tethering at mitochondria-associated ER membranes (MAMs)[106-108]. MAMs are essential to regulate key cellular functions regarding lipid and calcium homeostasis[106,109-111], as well as mitochondria dynamics and bioenergetics. In addition, it has been shown that MFN2, similar to GDAP1, is a tethering protein between mitochondria and lysosomes[67]. Studies to shed light on the pathophysiology of MFN2 mutations have been performed in nervous tissues and fibroblasts from patients, as well as in mice and motor neurons derived from induced pluripotent cells (iPSCs) obtained from fibroblasts. However, the results are quite controversial since some of the changes can only be observed in some studies but do not extend to all of them: for instance, there is still little agreement regarding whether there are alterations in MFN2 protein levels, in the respiratory chain capacity and oxidative phosphorylation, in mitochondrial membrane potential, or mtDNA content[101,112-114].
Most missense variants reside within MFN2 dynamin-GTPase domain[115,116], and there is recent evidence that a dominant-negative or gain-of-function effect may be responsible for the pathogenicity. Other evidence shows that some variants are responsible for mitochondrial hypofusion, while others can cause mitochondrial hyperfusion[117]. Moreover, as explained above, autosomal recessive and semidominant forms of CMT2A2 have been reported. This further illustrates the allelic heterogeneity of this condition[118].
Even with differences between studies, it is clear that there are mitochondrial abnormalities in MFN2 patients. Mitofusin-2 represents a key player in several mitochondrial activities: fusion, trafficking, turnover, and contacts with other organelles and the consequences of mutations in the MFN2 gene disbalance the appropriate mitochondrial shape, function, and distribution within the cell[119]. Alterations in mitochondrial transport and distribution likely cause a bioenergetics impairment, especially in highly metabolic cells, which is the case for neurons. These disrupted and affected processes lead to a loss of myelinated fibers and mitochondrial abnormalities, visible in nerve biopsies.
MPZ
Mutations in MPZ commonly result in autosomal dominant neuropathy and are estimated to account for 5% of cases of CMT[26]. In this gene, the phenotypical heterogeneity that can be found is remarkable, in regards to both the severity of the symptoms and the neurophysiological parameters.
Myelin protein zero (P0), which is part of the immunoglobulin gene superfamily, is a major peripheral protein that acts as a homophilic adhesion molecule and is crucial for compact myelin formation and maintenance in the PNS[120]. It is the most abundant myelin protein produced in myelinating Schwann cells. It has been related to CMT and can cause the three different types of the disease: demyelinating, axonal, and intermediate forms. Consequently, individuals carrying MPZ mutations have a variety of clinical phenotypes, from severe disease with early onset of weakness and sensory loss in the neonatal period associated with very low NCVs (Dejerine-Sottas syndrome) to a much milder disease with onset of symptoms in the fourth decade of life with minimal slowed NCVs (CMT2). However, variability also exists, since late-onset patients can be quite severe, too (even confined to a wheelchair), and onset can last until the eighth decade of life. In nerves, signs of demyelination/remyelination with myelin outfoldings and onion bulb formations are the most characteristics features.
The structure of P0 is divided into three different domains: extracellular, transmembrane, and cytoplasmatic domains. Interestingly, most of the described mutations can be found in the extracellular part, which is essential for establishing interactions. P0 forms homotetramers within the cell membrane: each homotetramer interacts in trans with a similar homotetramer on the opposing membrane surface. Furthermore, P0 tethers appose lipid bilayers together through its extracellular immunoglobulin-like domain[121]. This is the reason it was proposed as having a key role in myelination, as P0 holds together adjacent wraps of myelin membrane through these homotypic interactions. Not only the extracellular part is essential to establish these interactions, but also the cytoplasmatic domain, as has been shown in different studies[122]. P0 participates in an adhesion-mediated signal transduction cascade, which even further supports its essential role in myelination.
As mentioned above, from the clinical point of view, MPZ mutations have been linked to both infantile and late onset of the disease, and some reports have proposed that early-onset neuropathy is related to mutations that disrupt the tertiary structure of P0 and, thus, interfere with P0-mediated adhesion and myelin compaction, while late-onset neuropathy is caused by those that more subtly alter myelin structure, probably disrupting Schwann cell-axonal interactions[122]. This suggests that MPZ mutations that predominantly affect myelination during development cause early-onset disease, while those that affect axons cause late-onset disease.
Although the exact mechanism by which mutations in MPZ can lead to CMT disease is unknown, mutated P0 has been linked to the unfolded protein response (UPR), which would cause defects in translation rate, folding, and/or membrane insertion. Furthermore, misfolded protein toxicity or reduced amounts of P0 could be the etiology behind the phenotypic manifestations in patients carrying MPZ mutations[121]. Given that P0 is known to interact with lipid membrane surfaces[123,124], mutations within P0 could also have direct effects on the formation of mature compact myelin at a molecular level[125].
Although a complete genotype/phenotype relationship has not been established, it is clear that two groups of disease expressions can be delineated to classify the more than 200 different disease-causing mutations that have been reported in MPZ until today.
PMP22
The PMP22 gene has been linked to CMT since the beginning of the genetic findings in this disease, and to different phenotypic neuropathies since three mechanisms can lead to different disorders: (i) PMP22 overdose within CMT1A duplication causes the most common cause of the disease; (ii) point mutations of PMP22 may be the underlying cause of more severe and early-onset forms, CMT types 1A or 1E; and (iii) the monoallelic lack of PMP22 at 17p11.2 as a consequence of the 1.4 Mb deletion in the CMT1A locus[126] or point pathogenic variants[127] can lead to hereditary neuropathy with liability to pressure palsies (HNPP). As explained above, the overall prevalence of CMT is 1:2500, where 1:3800-12,500 corresponds to CMT1A[128-130].
The consequence of the 1.4 Mb CMT1A duplication or HNPP deletion is the expression of three copies or one copy, respectively, of dose-sensitive PMP22. Thus, gene dosage has been the proposed pathological mechanism, supported by the finding of increased protein and mRNA levels in CMT1A sural nerve biopsies. Since PMP22 gene is under tight regulation, small changes can be expected to cause defects in myelination and motor and sensory functions[131]. Furthermore, regulation of PMP22 expression also occurs during protein synthesis and translocation. From a histological point of view, PMP22 mutations are characterized by onion bulb formations (CMT1A duplication) or tomaculae (HNPP deletion). A reduction of myelinated fibers with signs of demyelination can be observed as a consequence of the functional impact on PMP22 protein.
PMP22 encodes a 22 kDa glycoprotein produced primarily in Schwann cells and comprises 5% of proteins of the PNS myelin[131]. It is expressed in the compact portion of essentially all myelinated fibers in peripheral nerves. It has been proposed as a key role player during Schwann cell growth and differentiation[130]. Nevertheless, the exact biological function is not clear yet. Proper folding and regulation of PMP22 are essential for myelinating Schwann cells. Additionally, PMP22 promotes the organization of membrane structure in compact myelin, plays a role in the maintenance of cholesterol homeostasis in Schwann cells, and is involved in adhesion and cell proliferation[131]. Finally, PMP22 point mutations are known to disrupt PMP22 plasma membrane trafficking, resulting in misfolded proteins that are targeted by the ER, associated with degradation for clearance. As this process is not 100% efficient, there is an accumulation of the misfolded protein, which produces cellular stress[131].
It is important to remark that, apart from MPZ mutations, PMP22 mutations can be the cause of Dejerine-Sottas syndrome, which occurs in the first two years of life[132].
Since this gene has been studied from the very beginning of genetic analysis in CMT, the knowledge around it has been sufficient to design several drugs and/or therapeutic approaches, with the objective of regulating its expression levels.
SH3TC2
SH3TC2 is associated with autosomal recessive demyelinating CMT type 4C (CMT4C)[133]. This type of CMT shows an early onset, characterized by unsteadiness, distal weakness, occasional cranial nerve involvement (hearing loss, pupillary abnormalities, and/or tongue atrophy), and foot and spinal deformities[134,135]. Traditionally, these features have been explained by the important sensory loss inherent to this type of CMT. However, the important vestibular impairment that was confirmed in previous depth characterizations of CMT4C patients should also be mentioned[134]. SH3TC2-related CMT cases are not distributed randomly, and several studies have shown substantial differences between countries[132,133,136]. Specifically, SH3TC2-causative variants have been proven to have a high prevalence in Spanish Gypsy cohorts of patients[137].
SH3TC2 encodes an effector molecule of Rab11, which is found in myelinating Schwann cells and expressed late during myelination, thus is essential in the maintenance of the structural integrity of peripheral nerve myelin sheaths[133]. To date, more than 70 mutations in this gene have been reported (dispersed throughout the protein) and proven to influence the peripheral nerve pathophysiology. Specifically, SH3TC2 participates in the endocytic pathway of cell traffic, and, therefore, it can be found in clathrin-coated vesicles (including the trans-Golgi-network, early and late endosomes, and specific domains of the plasma membrane). This localization is dependent on protein myristoylation and interactions, for which both its SH3 and TRP domains are essential[138]. Furthermore, evidence has been presented on its role in the Nrg1/ErbB signaling pathway during early postnatal development of the PNS, suggesting that CMT4C patient’s hypomyelination may be explained, at least in part, as a consequence of the dysregulation of this signaling pathway[139]. As a consequence of these molecular abnormalities, in biopsies of SH3TC2-patients, concentric Schwann cell proliferation with multiple small onion bulbs can be observed, with the involvement of unmyelinated fibers.
SORD
Sorbitol dehydrogenase gene (SORD) has recently been identified as a causative gene of recessive forms of hereditary neuropathy, both CMT type 2 and distal hereditary motor neuropathy (dHMN)[140]. This gene encodes a protein that acts as a key enzyme in the polyol pathway, which is an alternate route for sugar metabolism[141]. SORD catalyzes the interconversion between glucose and fructose via sorbitol, together with aldose reductase. Due to that, this protein is believed to be involved in the development of diabetic neuropathy[141].
The historical deficit in the diagnostic rate of SORD cases is a consequence of SORD2P, a SORD pseudogene. The homology between them is quite high, with an identity of 2295 out of 2320 bases, with three gaps. Pseudogenes are usually not actively transcribed or translated, and they can be recognized by the presence of nonsense mutations or a frameshift that interrupts the open reading frame[142].
Biallelic mutations in SORD gene were initially described in May 2020 and, until now, 14 mutations have been identified[141]. Among them, the most prevalent mutation is c.757delG, which is common to all the reported patients (either in homozygous or heterozygous states, except for one patient with dHMN). Among these 14 variants, most are frameshift or splicing mutations causing a loss of function of sorbitol dehydrogenase.
Some studies have found a complete loss of SORD protein and increased intracellular sorbitol levels in fibroblasts derived from patients. Some inhibitors of aldose reductase led to the rescue in Drosophila of synaptic degeneration and motor deficiency caused by SORD orthologs deletion[141]. However, the exact mechanism that produces the axonal damage that can be seen in SORD cases has not been elucidated yet. Mechanisms such as the increase of sorbitol levels, cellular osmolarity, oxidative stress, and the decrease of NADPH levels have been proposed as responsible for the phenotype[141]. Few data on nerve pathology are available. Recently, Chen et al. (2022) described the neuropathology in the sural nerve of a 25-year-old woman with dHMN phenotype, which confirmed slight changes, including thin myelin sheath fibers and separation from the myelin sheath in very few fibers[143]. These authors identified microvascular basement membrane thickening, something that has also related to diabetic neuropathy.
In a clinical setting, some efforts should be made to reanalyze those undiagnosed patients suffering from hereditary axonal neuropathies since it has been proven that SORD might play an important causative role. The recognition of the biallelic variants in this gene would help to increase the diagnostic rates of autosomal recessive and axonal types of neuropathies[144].
To check SORD-related CMT cases, it is important to consider three different aspects: (1) the clinical homogeneity of cases with an onset in the second or third life decade and axonal neuropathy with distal muscle weakness and atrophies; (2) the c.757del variant, which is common and facilitates genetic screening; and (3) if direct analysis of this gene is selected, the primers should be designed with special attention to discriminate SORD and SORD2P. Furthermore, in the case of NGS techniques, an optimization of the analysis pipeline should be considered, selecting proper parameters, especially regarding alignment settings[144].
Although we are far from understanding the exact mechanism of pathogenesis of SORD variants, there is no doubt that this gene is a key role player in axonal neuropathies. The analysis of its sequence should help to reduce the diagnostic deficit in cohorts of neuropathic patients.
CONCLUSION
Over the last decades, there have been significant advances in deciphering the genetic causes of CMT disease. The number of genes associated with this peripheral neuropathy increased markedly, and improvements have been made in the genetic diagnosis. To manage such a heterogeneous group of disorders, it is essential to make a correct classification of its different types and elucidate which is the particular mechanism that underlies the pathological effect of the variants in the associated genes. Thanks to the efforts made to characterize them, it is clear that CMT-associated genes are closely related to the formation, compaction, and maintenance of myelin, the neuronal soma, axon and cytoskeleton conservation, the axonal transport, and the mitochondrial dynamics. As a consequence, a degenerative axonal process can be expected as the final common pathway in genetic neuropathies, regardless of the metabolic/cytoplasmic/structural defect underlying the affectation of either the myelin, the axon, or the Schwann cell-axon structure. In the case of the most commonly involved genes, such as GDAP1, GJB1, HINT1, MFN2, MPZ, PMP22, SH3TC2, and SORD, a more in-depth analysis is carried out in this review to shed light on the pathophysiological mechanisms. This has proven to be useful in pinpointing molecular targets, thereby helping to identify and design therapeutical approaches. In conclusion, although nowadays we lack disease-modifying treatments, increasing knowledge on the molecular basis of CMT will help to reach them, which poses a promising future.
DECLARATIONS
Authors’ contributionsMade substantial contributions to concept, design and writing of the review: Estévez-Arias B, Carrera-Garcia L, Nascimento A, Cantarero L, Hoenicka J, Palau F
Performed critical revision of the manuscript for important intellectual content and approved the final version: Estévez-Arias B, Carrera-García L, Nascimento A, Cantarero L, Hoenicka J, Palau F
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Agencia Estatal de Investigación (Spanish Ministry of Science and Innovation) grant PID2020-114655RB-I00; the CIBERER-ACCI 2019-16; the Generalitat de Catalunya and European Regional Development Fund grants 2015 FEDER/S21, and 2017/SGR1308; the Fundación Isabel Gemio and the Torró Solidari-RAC1 i Torrons Vicens. The Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) is an initiative of the Instituto de Salud Carlos III, and the Department of Genetic Medicine of Sant Joan de Déu Children’s Hospital is part of the Centre Daniel Bravo de Diagnòstic i Recerca de Malalties Minoritàries.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
Supplementary MaterialsREFERENCES
1. Charcot JM, Marie P. Sur une forme particulière d’atrophie musculaire progressive souvent familiale débutant par les pieds et les jambes et atteignant plus tard les mains. Paris: Félix Alcan; 1886. pp. 97-138.
2. Tooth HH. The peroneal type of progressive muscular atrophy: a thesis for the degree of M.D. in the university of cambridge - digital collections - national library of medicine. Available from: https://collections.nlm.nih.gov/catalog/nlm:nlmuid-100899256-bk [Last accessed on 13 June 2022].
5. Barreto LC, Oliveira FS, Nunes PS, et al. Epidemiologic study of Charcot-Marie-Tooth disease: a systematic review. Neuroepidemiology 2016;46:157-65.
6. Combarros O, Calleja J, Polo JM, Berciano J. Prevalence of hereditary motor and sensory neuropathy in Cantabria. Acta Neurol Scand 1987;75:9-12.
7. Bird TD. Charcot-Marie-Tooth (CMT) hereditary neuropathy overview. 1998 Sep 28 [updated 2021 Sep 9]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®[Internet]. Seattle, WA: University of Washington, 1993-2022.
8. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980;103:259-80.
9. Werheid F, Azzedine H, Zwerenz E, et al. Underestimated associated features in CMT neuropathies: clinical indicators for the causative gene? Brain Behav 2016;6:e00451.
10. Anzalone CL, Nuhanovic S, Olund AP, Carlson ML. Cochlear implantation in Charcot-Marie-Tooth disease: case report and review of the literature. Case Rep Med 2018;2018:1760978.
11. Lerat J, Magdelaine C, Beauvais-Dzugan H, et al. A novel pathogenic variant of NEFL responsible for deafness associated with peripheral neuropathy discovered through next-generation sequencing and review of the literature. J Peripher Nerv Syst 2019;24:139-44.
12. Jani-Acsadi A, Ounpuu S, Pierz K, Acsadi G. Pediatric Charcot-Marie-Tooth disease. Pediatr Clin N Am 2015;62:767-86.
13. Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011;69:22-33.
14. Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol 2013;9:562-71.
15. Raeymaekers P, Timmerman V, Nelis E, et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). Neuromuscul Disord 1991;1:93-7.
16. Lupski JR, de Oca-luna RM, Slaugenhaupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991;66:219-32.
17. Gonzaga-Jauregui C, Harel T, Gambin T, et al. Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy . Cell Rep 2015;12:1169-83.
18. Blair IP, Nash J, Gordon MJ, Nicholson GA. Prevalence and origin of de novo duplications in Charcot-Marie-Tooth disease type 1A: first report of a de novo duplication with a maternal origin. Am J Hum Genet 1996;58:472-6.
19. Ouvrier R. What can we learn from the history of Charcot-Marie-Tooth disease? Dev Med Child Neurol 2010;52:405-6.
20. Magy L, Mathis S, Le Masson G, Goizet C, Tazir M, Vallat JM. Updating the classification of inherited neuropathies: Results of an international survey. Neurology 2018;90:e870-6.
21. Mathis S, Goizet C, Tazir M, et al. Charcot-Marie-Tooth diseases: an update and some new proposals for the classification. J Med Genet 2015;52:681-90.
22. Timmerman V, Strickland AV, Züchner S. Genetics of Charcot-Marie-Tooth (CMT) disease within the frame of the human genome project success. Genes 2014;5:13-32.
23. Høyer H, Braathen GJ, Busk ØL, et al. Genetic diagnosis of Charcot-Marie-Tooth disease in a population by next-generation sequencing. Biomed Res Int 2014;2014:210401.
24. Juárez P, Palau F. Neural and molecular features on Charcot-Marie-Tooth disease plasticity and therapy. Neural Plast 2012;2012:171636.
25. Cohen E, Bonne G, Rivier F, Hamroun D. The 2022 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul Disord 2021;31:1313-57.
26. Rossor AM, Evans MR, Reilly MM. A practical approach to the genetic neuropathies. Pract Neurol 2015;15:187-98.
27. Miller LJ, Saporta AS, Sottile SL, Siskind CE, Feely SM, Shy ME. Strategy for genetic testing in Charcot-Marie-disease. Acta Myol 2011;30:109-16.
28. Cortese A, Wilcox JE, Polke JM, et al. Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology 2020;94:e51-61.
29. Yubero D, Natera-de Benito D, Pijuan J, et al. The increasing impact of translational research in the molecular diagnostics of neuromuscular diseases. Int J Mol Sci 2021;22:4274.
30. Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.
31. Young P, De Jonghe P, Stögbauer F, Butterfass-Bahloul T. Treatment for Charcot-Marie-Tooth disease. Cochrane Database Syst Rev 2008;1:CD006052.
32. Sackley C, Disler PB, Turner-stokes L, et al. Rehabilitation interventions for foot drop in neuromuscular disease. Cochrane Database of Systematic Reviews. Cochrane Database Syst Rev 2009;8:CD003908.
33. Guillebastre B, Calmels P, Rougier PR. Assessment of appropriate ankle-foot orthoses models for patients with Charcot-Marie-Tooth disease. Am J Phys Med Rehabil 2011;90:619-27.
34. Jani-Acsadi A, Krajewski K, Shy ME. Charcot-Marie-Tooth neuropathies: diagnosis and management. Semin Neurol 2008;28:185-94.
35. Burns J, Crosbie J, Ouvrier R, Hunt A. Effective orthotic therapy for the painful cavus foot: a randomized controlled trial. J Am Podiatr Med Assoc 2006;96:205-11.
36. Piscosquito G, Reilly MM, Schenone A, et al. CMT-TRIAAL & CMT-TRAUK Group. Is overwork weakness relevant in Charcot-Marie-Tooth disease? J Neurol Neurosurg Psychiatry 2014;85:1354-8.
37. Beals TC, Nickisch F. Charcot-Marie-Tooth disease and the cavovarus foot. Foot Ankle Clin 2008;13:259-74.
39. Herrmann DN. Experimental therapeutics in hereditary neuropathies: the past, the present, and the future. Neurotherapeutics 2008;5:507-15.
40. zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann Neurol 2007;61:61-72.
41. Sahenk Z, Nagaraja HN, McCracken BS, et al. NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology 2005;65:681-9.
42. Passage E, Norreel JC, Noack-Fraissignes P, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med 2004;10:396-401.
44. Gess B, Baets J, De Jonghe P, Reilly MM, Pareyson D, Young P. Ascorbic acid for the treatment of Charcot-Marie-Tooth disease. Cochrane Database Syst Rev 2015;12:CD011952.
45. Sereda MW, Meyer zu Hörste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med 2003;9:1533-7.
46. Attarian S, Young P, Brannagan TH, et al. A double-blind, placebo-controlled, randomized trial of PXT3003 for the treatment of Charcot-Marie-Tooth type 1A. Orphanet J Rare Dis 2021;16:433.
47. d'Ydewalle C, Krishnan J, Chiheb DM, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 2011;17:968-74.
48. Adalbert R, Kaieda A, Antoniou C, et al. Novel HDAC6 Inhibitors increase tubulin acetylation and rescue axonal transport of mitochondria in a model of Charcot-Marie-Tooth type 2F. ACS Chem Neurosci 2020;11:258-67.
49. Picci C, Wong VSC, Costa CJ, et al. HDAC6 inhibition promotes α-tubulin acetylation and ameliorates CMT2A peripheral neuropathy in mice. Exp Neurol 2020;328:113281.
50. Pisciotta C, Saveri P, Pareyson D. Challenges in treating Charcot-Marie-Tooth disease and related neuropathies: current management and future perspectives. Brain Sci 2021;11:1447.
51. Rossaert E, Van Den Bosch L. HDAC6 inhibitors: translating genetic and molecular insights into a therapy for axonal CMT. Brain Res 2020;1733:146692.
52. Nuevo-Tapioles C, Santacatterina F, Sánchez-Garrido B, et al. Effective therapeutic strategies in a preclinical mouse model of Charcot-Marie-Tooth disease. Hum Mol Genet 2021;30:2441-55.
53. Kagiava A, Karaiskos C, Richter J, et al. AAV9-mediated schwann cell-targeted gene therapy rescues a model of demyelinating neuropathy. Gene Ther 2021;28:659-75.
54. Marco A, Cuesta A, Pedrola L, Palau F, Marín I. Evolutionary and structural analyses of GDAP1, involved in Charcot-Marie-Tooth disease, characterize a novel class of glutathione transferase-related genes. Mol Biol Evol 2004;21:176-87.
55. Huber N, Bieniossek C, Wagner KM, et al. Glutathione-conjugating and membrane-remodeling activity of GDAP1 relies on amphipathic C-terminal domain. Sci Rep 2016;6:36930.
56. Chen CX, Li JQ, Dong HL, Liu GL, Bai G, Wu ZY. Identification and functional characterization of novel GDAP1 variants in Chinese patients with Charcot-Marie-Tooth disease. Ann Clin Transl Neurol 2020;7:2381-92.
57. Niemann A, Ruegg M, La Padula V, Schenone A, Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol 2005;170:1067-78.
58. Pedrola L, Espert A, Valdés-Sánchez T, et al. Cell expression of GDAP1 in the nervous system and pathogenesis of Charcot-Marie-Tooth type 4A disease. J Cell Mol Med 2008;12:679-89.
60. Azzedine H, Ruberg M, Ente D, et al. Variability of disease progression in a family with autosomal recessive CMT associated with a S194X and new R310Q mutation in the GDAP1 gene. Neuromuscul Disord 2003;13:341-6.
61. Manganelli F, Pisciotta C, Nolano M, et al. A novel autosomal dominant GDAP1 mutation in an Italian CMT2 family. J Peripher Nerv Syst 2012;17:351-5.
62. Cantarero L, Juárez-Escoto E, Civera-Tregón A, et al. Mitochondria-lysosome membrane contacts are defective in GDAP1-related Charcot-Marie-Tooth disease. Hum Mol Genet 2021;29:3589-605.
63. Sevilla T, Cuesta A, Chumillas MJ, et al. Clinical, electrophysiological and morphological findings of Charcot-Marie-Tooth neuropathy with vocal cord palsy and mutations in the GDAP1 gene. Brain 2003;126:2023-33.
64. Niemann A, Wagner KM, Ruegg M, Suter U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol Dis 2009;36:509-20.
65. Noack R, Frede S, Albrecht P, et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum Mol Genet 2012;21:150-62.
66. Pla-Martín D, Rueda CB, Estela A, et al. Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol Dis 2013;55:140-51.
67. Pijuan J, Cantarero L, Natera-de Benito D, et al. Mitochondrial dynamics and mitocondria-lysosome contacts in neurogenetic diseases. Front Neurosci 2022;16:784880.
68. Cassereau J, Chevrollier A, Gueguen N, et al. Mitochondrial complex I deficiency in GDAP1-related autosomal dominant Charcot-Marie-Tooth disease (CMT2K). Neurogenetics 2009;10:145-50.
69. García-Sobrino T, Blanco-Arias P, Palau F, et al. Phenotypical features of a new dominant GDAP1 pathogenic variant (p.R226del) in axonal Charcot-Marie-Tooth disease. Neuromuscul Disord 2017;27:667-72.
70. González-Sánchez P, Pla-Martín D, Martínez-Valero P, et al. CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca2+ entry-stimulated respiration. Sci Rep 2017;7:42993.
71. Cassereau J, Chevrollier A, Codron P, et al. Oxidative stress contributes differentially to the pathophysiology of Charcot-Marie-Tooth disease type 2K. Exp Neurol 2020;323:113069.
72. Kleopa KA, Abrams CK, Scherer SS. How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res 2012;1487:198-205.
73. Latour P, Gonnaud PM, Ollagnon E, et al. SIMPLE mutation analysis in dominant demyelinating Charcot-Marie-Tooth disease: three novel mutations. J Peripher Nerv Syst 2006;11:148-55.
75. Abrams CK. GJB1 disorders: Charcot-Marie-Tooth Neuropathy (CMT1X) and central nervous system phenotypes. 1998 Jun 18 [updated 2020 Feb 20]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, 1993-2022.
76. Siskind CE, Murphy SM, Ovens R, Polke J, Reilly MM, Shy ME. Phenotype expression in women with CMT1X. J Peripher Nerv Syst 2011;16:102-7.
77. Dubourg O, Tardieu S, Birouk N, et al. Clinical, electrophysiological and molecular genetic characteristics of 93 patients with X-linked Charcot-Marie-Tooth disease. Brain 2001;124:1958-67.
78. Tsai PC, Chen CH, Liu AB, et al. Mutational analysis of the 5' non-coding region of GJB1 in a Taiwanese cohort with Charcot-Marie-Tooth neuropathy. J Neurol Sci 2013;332:51-5.
79. Hahn AF, Ainsworth PJ, Bolton CF, Bilbao JM, Vallat JM. Pathological findings in the x-linked form of Charcot-Marie-Tooth disease: a morphometric and ultrastructural analysis. Acta Neuropathol 2001;101:129-39.
80. Abrams CK, Freidin M. GJB1-associated X-linked Charcot-Marie-Tooth disease, a disorder affecting the central and peripheral nervous systems. Cell Tissue Res 2015;360:659-73.
81. Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 2002;22:6458-70.
82. Balice-Gordon RJ, Bone LJ, Scherer SS. Functional gap junctions in the schwann cell myelin sheath. J Cell Biol 1998;142:1095-104.
83. Chandross KJ, Kessler JA, Cohen RI, et al. Altered connexin expression after peripheral nerve injury. Mol Cell Neurosci 1996;7:501-18.
84. Scherer S, Deschenes S, Xu Y, Grinspan J, Fischbeck K, Paul D. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci 1995;15:8281-94.
85. Sutor B, Schmolke C, Teubner B, Schirmer C, Willecke K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb Cortex 2000;10:684-97.
86. Beauvais K, Furby A, Latour P. Clinical, electrophysiological and molecular genetic studies in a family with X-linked dominant Charcot-Marie-Tooth neuropathy presenting a novel mutation in GJB1 Promoter and a rare polymorphism in LITAF/SIMPLE. Neuromuscul Disord 2006;16:14-8.
87. Houlden H, Girard M, Cockerell C, et al. Connexin 32 promoter P2 mutations: a mechanism of peripheral nerve dysfunction. Ann Neurol 2004;56:730-4.
88. Gonzaga-Jauregui C, Zhang F, Towne CF, Batish SD, Lupski JR. GJB1/Connexin 32 whole gene deletions in patients with X-linked Charcot-Marie-Tooth disease. Neurogenetics 2010;11:465-70.
89. Bicego M, Morassutto S, Hernandez VH, et al. Selective defects in channel permeability associated with Cx32 mutations causing X-linked Charcot-Marie-Tooth disease. Neurobiol Dis 2006;21:607-17.
90. Abrams CK, Islam M, Mahmoud R, Kwon T, Bargiello TA, Freidin MM. Functional requirement for a highly conserved charged residue at position 75 in the gap junction protein connexin 32. J Biol Chem 2013;288:3609-19.
91. Jeng LJ, Balice-Gordon RJ, Messing A, Fischbeck KH, Scherer SS. The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol Cell Neurosci 2006;32:283-98.
92. Zimoń M, Baets J, Almeida-Souza L, et al. Loss-of-function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nat Genet 2012;44:1080-3.
93. Peeters K, Chamova T, Tournev I, Jordanova A. Axonal neuropathy with neuromyotonia: there is a HINT. Brain 2017;140:868-77.
94. Rauchenzauner M, Frühwirth M, Hecht M, Kofler M, Witsch-Baumgartner M, Fauth C. A Novel variant in the HINT1 gene in a girl with autosomal recessive axonal neuropathy with neuromyotonia: thorough neurological examination gives the clue. Neuropediatrics 2016;47:119-22.
95. Caetano JS, Costa C, Baets J, et al. Autosomal recessive axonal neuropathy with neuromyotonia: a rare entity. Pediatr Neurol 2014;50:104-7.
96. Laššuthová P, Brožková DŠ, Krůtová M, et al. Mutations in HINT1 are one of the most frequent causes of hereditary neuropathy among Czech patients and neuromyotonia is rather an underdiagnosed symptom. Neurogenetics 2015;16:43-54.
97. Kontogeorgiou Z, Voudommatis C, Kartanou C, et al. HINT1-related neuropathy in Greek patients with Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2021;26:444-8.
98. Chou TF, Sham YY, Wagner CR. Impact of the C-terminal loop of histidine triad nucleotide binding protein1 (Hint1) on substrate specificity. Biochemistry 2007;46:13074-9.
99. Ouvrier R, Mcleod J, Morgan G, Wise G, Conchin T. Hereditary motor and sensory neuropathy of neuronal type with onset in early childhood. J Neurol Sci 1981;51:181-97.
100. Lee M, Park CH, Chung HK, et al. Cerebral white matter abnormalities in patients with charcot-marie-tooth disease. Ann Neurol 2017;81:147-51.
101. Larrea D, Pera M, Gonnelli A, et al. MFN2 mutations in Charcot-Marie-Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum Mol Genet 2019;28:1782-800.
102. Pipis M, Feely SME, Polke JM, et al. Natural history of Charcot-Marie-Tooth disease type 2A: a large international multicentre study. Brain 2020;143:3589-602.
103. Bombelli F, Stojkovic T, Dubourg O, et al. Charcot-Marie-Tooth disease type 2A: from typical to rare phenotypic and genotypic features. JAMA Neurol 2014;71:1036-42.
104. Feely SM, Laura M, Siskind CE, et al. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 2011;76:1690-6.
105. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012;337:1062-5.
106. Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008;456:605-10.
107. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004;305:858-62.
108. Chen H, Chan DC. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum Mol Genet 2009;18:R169-76.
109. Leal NS, Schreiner B, Pinho CM, et al. Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production. J Cell Mol Med 2016;20:1686-95.
110. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci USA 2015;112:E2174-81.
111. Merkwirth C, Langer T. Mitofusin 2 builds a bridge between ER and mitochondria. Cell 2008;135:1165-7.
112. Vielhaber S, Debska-Vielhaber G, Peeva V, et al. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol 2013;125:245-56.
113. Amiott EA, Lott P, Soto J, et al. Mitochondrial fusion and function in Charcot-Marie-Tooth type 2A patient fibroblasts with mitofusin 2 mutations. Exp Neurol 2008;211:115-27.
114. Loiseau D, Chevrollier A, Verny C, et al. Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A disease. Ann Neurol 2007;61:315-23.
115. Chandhok G, Lazarou M, Neumann B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol Rev Camb Philos Soc 2018;93:933-49.
116. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010;11:872-84.
117. El Fissi N, Rojo M, Aouane A, et al. Mitofusin gain and loss of function drive pathogenesis in. Drosophila 2018;19:e45241.
118. Tomaselli PJ, Kapoor M, Cortese A, Polke JM, Rossor AM, Reilly MM. Severe cognitive impairment in a patient with CMT2A. J Peripher Nerv Syst 2018;23:147-8.
119. Filadi R, Pendin D, Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis 2018;9:330.
120. Otani Y, Ohno N, Cui J, Yamaguchi Y, Baba H. Upregulation of large myelin protein zero leads to Charcot-Marie-Tooth disease-like neuropathy in mice. Commun Biol 2020;3:121.
121. Jerath NU, Shy ME. Hereditary motor and sensory neuropathies: Understanding molecular pathogenesis could lead to future treatment strategies. Biochim Biophys Acta 2015;1852:667-78.
122. Shy ME, Jáni A, Krajewski K, et al. Phenotypic clustering in MPZ mutations. Brain 2004;127:371-84.
123. Raasakka A, Ruskamo S, Kowal J, et al. Molecular structure and function of myelin protein P0 in membrane stacking. Sci Rep 2019;9:642.
124. Luo X, Sharma D, Inouye H, et al. Cytoplasmic domain of human myelin protein zero likely folded as beta-structure in compact myelin. Biophys J 2007;92:1585-97.
125. Bai Y, Wu X, Brennan KM, et al. Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Ann Clin Transl Neurol 2018;5:445-55.
126. Chance PF, Alderson MK, Leppig KA, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993;72:143-51.
127. Bort S, Nelis E, Timmerman V, et al. Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum Genet 1997;99:746-54.
128. Braathen GJ, Sand JC, Lobato A, Høyer H, Russell MB. Genetic epidemiology of Charcot-Marie-Tooth in the general population. Eur J Neurol 2011;18:39-48.
129. Karadima G, Floroskufi P, Koutsis G, Vassilopoulos D, Panas M. Mutational analysis of PMP22, GJB1 and MPZ in Greek Charcot-Marie-Tooth type 1 neuropathy patients. Clin Genet 2011;80:497-9.
130. Paassen BW, van der Kooi AJ, van Spaendonck-Zwarts KY, Verhamme C, Baas F, de Visser M. PMP22 related neuropathies: Charcot-Marie-Tooth disease type 1A and Hereditary Neuropathy with liability to Pressure Palsies. Orphanet J Rare Dis 2014;9:38.
131. Boutary S, Echaniz-Laguna A, Adams D, et al. Treating PMP22 gene duplication-related Charcot-Marie-Tooth disease: the past, the present and the future. Transl Res 2021;227:100-11.
132. Li J, Parker B, Martyn C, Natarajan C, Guo J. The PMP22 gene and its related diseases. Mol Neurobiol 2013;47:673-98.
133. Yuan JH, Hashiguchi A, Okamoto Y, et al. Clinical and mutational spectrum of Japanese patients with recessive variants in SH3TC2. J Hum Genet 2018;63:281-7.
134. Pérez-Garrigues H, Sivera R, Vílchez JJ, Espinós C, Palau F, Sevilla T. Vestibular impairment in Charcot-Marie-Tooth disease type 4C. J Neurol Neurosurg Psychiatry 2014;85:824-7.
135. Claramunt R, Sevilla T, Lupo V, et al. The p.R1109X mutation in SH3TC2 gene is predominant in Spanish Gypsies with Charcot-Marie-Tooth disease type 4. Clin Genet 2007;71:343-9.
136. Laššuthová P, Mazanec R, Vondráček P, et al. High frequency of SH3TC2 mutations in Czech HMSN I patients. Clin Genet 2011;80:334-45.
137. Azzedine H, Salih MA. SH3TC2-related hereditary motor and sensory neuropathy. 2008 Mar 31 [updated 2021 Mar 11]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, 1993-2022.
138. Lupo V, Galindo MI, Martínez-Rubio D, et al. Missense mutations in the SH3TC2 protein causing Charcot-Marie-Tooth disease type 4C affect its localization in the plasma membrane and endocytic pathway. Hum Mol Genet 2009;18:4603-14.
139. Gouttenoire EA, Lupo V, Calpena E, et al. Sh3tc2 deficiency affects neuregulin-1/ErbB signaling. Glia 2013;61:1041-51.
140. Cortese A, Zhu Y, Rebelo AP, et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat Genet 2020;52:473-81.
141. Dong HL, Li JQ, Liu GL, Yu H, Wu ZY. Biallelic SORD pathogenic variants cause Chinese patients with distal hereditary motor neuropathy. NPJ Genom Med 2021;6:1.
142. Liu X, He J, Yilihamu M, Duan X, Fan D. Clinical and genetic features of biallelic mutations in. SORD 2021;12:733926.
143. Chen B, Zhang Z, Zhang C, et al. Clinical and pathological study of SORD-related distal motor neuropathy caused by novel compound heterozygous mutations in a Chinese patient. Clin Neurol Neurosurg 2022;213:107118.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Estévez-Arias B, Carrera-García L, Nascimento A, Cantarero L, Hoenicka J, Palau F. Genetic approaches and pathogenic pathways in the clinical management of Charcot-Marie-Tooth disease. J Transl Genet Genom 2022;6:333-52. http://dx.doi.org/10.20517/jtgg.2022.04
AMA Style
Estévez-Arias B, Carrera-García L, Nascimento A, Cantarero L, Hoenicka J, Palau F. Genetic approaches and pathogenic pathways in the clinical management of Charcot-Marie-Tooth disease. Journal of Translational Genetics and Genomics. 2022; 6(3): 333-52. http://dx.doi.org/10.20517/jtgg.2022.04
Chicago/Turabian Style
Estévez-Arias, Berta, Laura Carrera-García, Andrés Nascimento, Lara Cantarero, Janet Hoenicka, Francesc Palau. 2022. "Genetic approaches and pathogenic pathways in the clinical management of Charcot-Marie-Tooth disease" Journal of Translational Genetics and Genomics. 6, no.3: 333-52. http://dx.doi.org/10.20517/jtgg.2022.04
ACS Style
Estévez-Arias, B.; Carrera-García L.; Nascimento A.; Cantarero L.; Hoenicka J.; Palau F. Genetic approaches and pathogenic pathways in the clinical management of Charcot-Marie-Tooth disease. J. Transl. Genet. Genom. 2022, 6, 333-52. http://dx.doi.org/10.20517/jtgg.2022.04
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 50 clicks
Cite This Article 50 clicks

 Like This Article 36
likes
Like This Article 36
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.