
The many roles of the Alzheimer-associated gene PM20D1
 0
0
Abstract
PM20D1 is a little studied enzyme until recently, belonging to the mammalian M20 peptidase family, which catalyzes both the synthesis and hydrolysis of N-acyl amino acids (NAAs). NAAs are bioactive lipids biosynthesized from free fatty acids and free amino acids. These molecules have been associated with many biological functions; however, most of the biochemical mechanisms have not yet been described. The best-known biochemical mechanism is the one involved in thermogenesis, which also has implications for reactive oxygen species levels and cell preservation. In the last few years, genetic variation in PM20D1, as well as changes in its methylation and expression levels, have been reported to be associated with several disease phenotypes, including Alzheimer’s disease. In this review, we explore the current knowledge regarding the PM20D1 gene, including aspects such as its biology, potential functions, regulation of its expression, and role in different phenotypes such as Alzheimer’s disease, obesity, Parkinson’s disease, and several other disorders.
Keywords
INTRODUCTION
In the last decades, a large amount of evidence regarding genetic risk variants for most common disorders has been generated mostly through genome-wide association studies (GWAS)[1]. However, less attention has been focused on the changes in methylation and expression associated with different phenotypes. This has changed in recent years with the increased use of whole-genome methylation and RNA sequencing techniques[2,3] (in addition to the long-existing expression microarray analyses). Some of the challenges faced by studies dealing with methylation and expression include their usually dramatically smaller sample sizes compared to GWAS, and that they often focus on either rather than both methylation and expression at the same time.
Such methylation studies have cast a spotlight on an until then little-studied gene, PM20D1, which encodes an enzyme belonging to the M20 peptidase family[4]. There is growing evidence that differential methylation in this gene is associated with several heterogeneous disease phenotypes [Table 1]. Additionally, it was discovered that genetic variants close to but not inside the gene can influence methylation levels at the gene’s promoter[5-8], and this methylation correlates with expression[6]. It has been proposed that these variants had not been previously found in GWAS because the PM20D1 region is not well represented and is in low linkage disequilibrium with the SNPs included in the common microarrays used in GWAS[6].
Phenotypes with reported differential methylation between cases and controls in human tissues and variants associated with PM20D1 promoter methylation
| Phenotype | Methylation status in cases | Tissue | mQTL variants* | References |
| Alzheimer | Hypermethylation (in advanced disease) | Brain prefrontal cortex[6,7], immortalized B cells[6], peripheral blood[7,8] | rs1172198[5] rs708727[5-8] rs823082[5] rs823088[5] rs1361754[5] rs960603[5,6] | [6-8] |
| Hypomethylation/overexpression (close to diagnosis and in early disease) | Postmortem brain[5], brain prefrontal cortex[6,7], immortalized B cells[6], peripheral blood[7,45] | [5-7,45] | ||
| Obesity (BMI) | Hypomethylation | Adipose tissue[36], peripheral blood[52] | rs823080[36] | [36,52] |
| Hypermethylation | Peripheral blood | - | [47] | |
| Parkinson | Hypermethylation in one study, inconclusive evidence in the other | Peripheral blood | rs823114[83] | [82,83] |
| Asthma | Hypomethylation | Peripheral blood | - | [86] |
| Respiratory allergy | Hypermethylation | Peripheral blood and saliva | - | [87] |
| Food allergy | Hypomethylation | Peripheral blood | - | [88] |
| Psoriasis | Hypermethylation | Peripheral blood | - | [89] |
| Multiple sclerosis | Direction of change not stated | Peripheral blood | - | [90] |
| Child abuse | Hypermethylation | Peripheral blood | - | [91] |
| Familial hypercholesterolemia | Direction of change not stated | Peripheral blood | - | [94] |
| Stroke | Hypermethylation | Peripheral blood | - | [96] |
| Covid severity | Direction of change not stated | Peripheral blood | - | [98] |
| Lung cancer | Direction of change not stated | Lung, bronchus | - | [99] |
| Hepatocellular carcinoma | Hypermethylation | Liver | - | [100] |
| Acute myeloid leukemia | Hypermethylation | Bone marrow mesenchymal stem cells | - | [101] |
| Chronic postsurgical pain | Hypermethylation | Peripheral blood | rs4951261 rs960603 rs708723 rs823114 rs11240547 rs2793374 | [102] |
In this review, we explore the current knowledge regarding the PM20D1 gene. First, we focus on what is known about its biology, potential functions, and regulation of its expression. We then review the evidence supporting the involvement of PM20D1 in different phenotypes such as Alzheimer’s disease (AD), obesity, Parkinson’s disease (PD), and several other disorders.
BIOLOGY
PM20D1 belongs to the mammalian M20 peptidase family[4]. The main function of this secreted enzyme is the synthesis and hydrolysis of N-acyl amino acids (NAAs)[9,10] by catalyzing the biosynthesis of NAAs from free fatty acids and free amino acids, as well as the reverse hydrolysis reaction[9]. This gene is expressed in several tissues such as the liver, bladder, brain, large intestine, pancreas, kidney, and heart of mice[11]. In humans, it shows a notably high expression in pancreas and skin, but is also expressed in many other tissues[12]. In the case of the brain, expression occurs across all brain regions[12,13] and cell types[14,15]. This peptidase circulates through the bloodstream in tight association with low- and high-density lipoproteins. These lipoproteins work as coactivators in PM20D1 activity and as biosynthesis sites of the NAAs[16].
NAAs are bioactive lipids composed of a fatty acyl chain linked to an amino acid by an amide bond[17]. NAAs circulate through the bloodstream, with albumin as a physiologic N-acyl amino acid carrier which confers resistance to hydrolytic degradation by spatially segregating N-acyl amino acids away from their site of biosynthesis. Albumin also helps to maintain equilibrium by acting as a buffer between bound inactive and free active NAAs[16]. Many NAAs identified in mammals have putative roles associated with different physiological processes. Some of the biological activities associated with NAAs are vasodilation[18], neuroprotection[19], and pain sensation[20,21]. However, many of the biochemical mechanisms that explain the role of the NAAs are still unknown[22].
Some NAAs have been described as thermogenic, for example, N-acyl-phenylalanines and N-acyl-leucines, which regulate energy metabolism[9,10,23]. The levels of these NAAs are physiologically increased after cold exposure and cause an uncoupling of mitochondrial respiration in different peripheral tissues by directly interacting with mitochondrial proteins[10,24,25]. Mitochondrial uncoupling occurs when ATP is not produced through electron transport[26], but redox energy is released in the form of heat, since protons are lost through the inner mitochondrial membrane[27]. This uncoupling activity is mostly limited to NAAs with neutral amino acid head groups and desaturated fatty acyl chains of medium length[23]. In mice, NAAs produce more energy expenditure and improve glucose homeostasis[9].
This thermogenic mitochondrial respiration uncoupling mechanism, activated by PM20D1 through direct binding of NAAs to mitochondria, is an independent alternative to uncoupling protein 1 (UCP1)[9,25]. UCP1 can be found in brown and beige adipocytes and can dissipate energy in the form of heat[28]. In the alternative mechanism, PM20D1 is expressed mainly from adipocytes which express UCP1, resulting in the generation of NAAs. These NAAs then promote respiration uncoupling both in the UCP1/PM20D1-expressing adipocytes and neighboring adipocytes lacking UCP1, which confirms that the two mechanisms are independent[9]. Furthermore, in vitro evidence exists that NAAs can induce uncoupled respiration in unrelated cell types that completely lack UCP1. Therefore, through this PM20D1-dependent thermogenic mechanism, respiration uncoupling (resulting in glucose degradation without production of ATP) can occur in cells that are not specialized in dissipating chemical energy as heat[9].
Some benefits associated with mitochondrial uncoupling are the reduction of the proton motive force (Δp), which causes a local decrease in oxygen concentration and a reduction of reactive oxygen species (ROS) products[29-31]. Δp plays a very important role in the entry of certain proteins and calcium into the mitochondria[29]. In addition, the activation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (mPTP) involved in the initiation of apoptosis is reduced[29,32]. Additionally, mitochondrial uncoupling promotes neuronal survival since these cells are highly oxidative and generate high levels of ROS[33]. Therefore, PM20D1 may be involved in a method of mitochondrial uncoupling (through NAAs) that modulates ROS levels and improves neuronal survival, potentially playing a role in neurodegenerative diseases[5].
GENE EXPRESSION
Multiple recent studies have shown that PM20D1 expression is, at least in part, genetically determined. Six SNPs (rs1172198, rs708727, rs823082, rs823088, rs1361754, and rs960603), which are located downstream of the gene, have been shown to be associated with both PM20D1 methylation and expression levels and are thus considered methylation QTLs (mQTLs) as well as expression QTLs (eQTLs)[5-8,34]. These SNPs constitute a haplotype which plays a role in the methylation of the PM20D1 promoter and its expression. As would be expected, the haplotype associated with higher methylation of the promoter is also associated with reduced expression of the gene. Sanchez-Mut et al. (2018) proposed that the genomic region where this haplotype is located acts as a regulatory region and induces repression through interaction with PM20D1’s promoter via a CTCF-mediated chromatin loop[6].
There are other genes on chromosome 1q in partial linkage disequilibrium with PM20D1. A study found a correlation between the genotype at these SNPs and expression in different tissues for other genes in the region besides PM20D1, specifically NUCKS1, RAB7L1, and SLC41A1[35]. Given that an association between the genotype at these SNPs (individually or in combination) and different disorders has been reported[5-8,36,37], the possibility of genotype-dependent expression for all four genes would raise the question of which gene is involved in a particular phenotype. To explore this, Sanchez-Mut et al. used a well-characterized sample of human brains to explore the DNA methylation levels in these four genes and found a strong correlation between genetic background (SNP genotypes) and CpG methylation only for PM20D1, as well as a slight correlation for SLC41A1 for only two of the SNPs[5]. Then, they looked at expression levels and found a significant correlation between genetic background and expression levels only for the PM20D1 gene, which they also observed in mice. They found a non-significant correlation trend between genotype and expression for SLC41A1 in the human samples, in the same direction as in PM20D1. Therefore, in the brain, the genotype at these SNPs seems to strongly influence methylation and expression levels of PM20D1 and possibly a lower degree for SLC41A1, but no effect on NUCKS1 and RAB7L1.
However, SLC41A1 and PM20D1 are differentially regulated by AD-related stressors, with only PM20D1 being upregulated by both amyloid-β and reactive oxygen species and only PM20D1 being neuroprotective when overexpressed in cell and primary cultures[5]. Therefore, at least in the case of AD, the evidence suggests that, from this region, PM20D1 is the main gene whose expression level is related to the phenotype.
From these and other studies, SNP rs708727 has emerged as the most significantly associated with PM20D1 methylation and expression levels in the brain and other tissues. For the other SNPs that constitute a haplotype with rs708727, the level of association is less significant and varies between studies[5-8,35,38-41]. SNP rs708727 is a coding variant in the SLC41A1 gene (not PM20D1) that results in the synonymous substitution p.Asn252Asn (NM_173854). The hypermethylation-associated allele is the A allele, with frequencies that vary between 0.3% in East Asians to over 44% in Finnish Europeans, according to gnomAD[42]. In the presence of the A allele, PM20D1’s promoter is hypermethylated, with the result that there is no transcription. Methylation levels are much lower when the G allele is present, and transcription can occur[5-8] [Figure 1]. Therefore, the effect of outside factors on expression levels in the gene (e.g., oxidative damage-induced hypomethylation) can be seen mostly for chromosomes with the G allele[7].
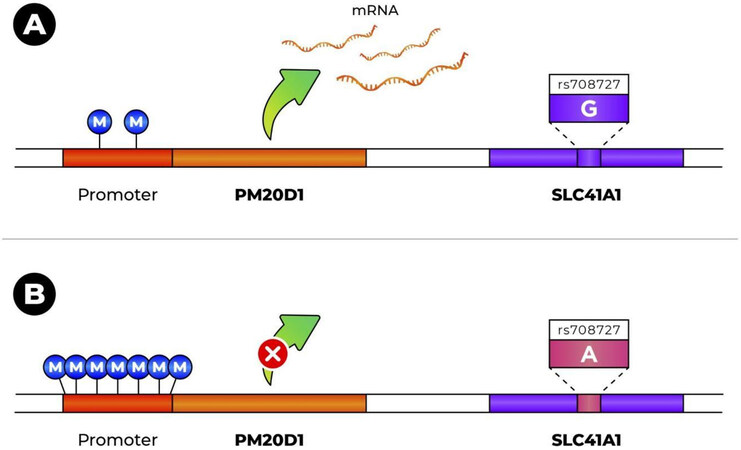
Figure 1. Effect of the rs708727 genotype on promoter methylation and expression level of PM20D1: (A) presence of G allele induces PM20D1 promoter hypomethylation and allows transcription; and (B) presence of A allele induces PM20D1 promoter hypermethylation and prevents its transcription.
This level of regulation of PM20D1 expression has been described as an on-off switch that acts in all human tissues. Additionally, specifically in adipocytes, a variant near the gene is involved in expression regulation by the peroxisome proliferator-activated receptor γ (PPARγ) transcription factor[36] (see the section on obesity).
The role of rs708727 as an mQTL appears to be consistent across populations of different ancestries. Differential methylation in PM20D1’s promoter that has been reported among Caucasian American, African- American, and Han Chinese American individuals can be explained by differences in allele frequencies at the rs708727 locus[42,43]. Additionally, the association between rs708727 genotype and PM20D1 methylation has also been reported in a sample of Costa Rican women[8].
ALZHEIMER’S
In 2018, in a very thorough study, Sánchez-Mut et al. reported for the first time an association of methylation status of the PM20D1 promoter with Alzheimer’s disease[6]. They found the promoter to be consistently hypermethylated in individuals with advanced-stage AD. The gene’s promoter had been previously shown to be differentially methylated between human populations of different ethnic origins[43]. The authors proceeded to identify several SNPs that correlated in an allele-dose-dependent manner with PM20D1 methylation; rs708727 has been found in later studies to show the most significant association and acts as an mQTL[7,8] (as described in the previous section). Moreover, they found that PM20D1 expression was inversely correlated with the methylation of its promoter. Variant rs708727 and several SNPs in linkage disequilibrium with it have been previously described as eQTLs for PM20D1[34]. Additionally, using peripheral blood, another recent study detected an association of a differentially methylated region in PM20D1 with the rate of cognitive decline in AD, as well as with the transition from cognitively healthy to the presence of cognitive impairment[44].
There is also functional evidence linking PM20D1 to AD. In cell culture, PM20D1 expression increased after treatment with neurotoxic insults related to AD, such as reactive oxygen species (ROS) and amyloid-β[5,6]. Additionally, in a mouse model with AD-related pathologies (APP/PS1), PM20D1 expression was higher in the frontal cortex at symptomatic stages than in pre-symptomatic stages and control mice[6]. Finally, in vitro overexpression of PM20D1 has been shown to decrease ROS-induced cell death and levels of amyloid-β in vitro, as well as reduce the amount of amyloid plaque and improve cognitive performance in mice[5,6]. These results suggest a neuroprotective role of PM20D1, but at first glance seem to be in contradiction with the hypermethylation (and presumed reduced expression) reported in individuals with advanced AD[6,8].
In the meantime, a mechanistic model has been proposed for the role of PM20D1 in AD[5-7], which explains the apparent contradiction. In individuals with hypermethylated PM20D1 (induced in part by the A allele rs708727), there is no transcription and therefore no PM20D1-mediated-protection against damage. On the other hand, in individuals without the methylation-inducing allele in rs708727 (i.e., individuals with the GG genotype), expression is increased in the presence of AD-related stress in order to reduce ROS-induced cell death, reduce Aβ levels, and prevent cognitive damage. Studies with longitudinal data have shown that hypomethylation occurs before the symptomatic onset of the disease, and therefore pre-diagnosis[7,45], potentially to increase gene expression and generate protection from damage. Then, a gradual increase in methylation is seen during disease progression in individuals with AD, leading to a decrease in gene expression [Figure 2]. This explains the previously reported hypermethylation in advanced AD[6,8]. In one study, thanks to the prospective follow-up of individuals who converted from not affected to presenting AD, the authors identified the turning point in methylation level at 78-79 years of age[7]. They also found a higher risk for hypermethylation of the PM20D1 promoter for females compared to male individuals.
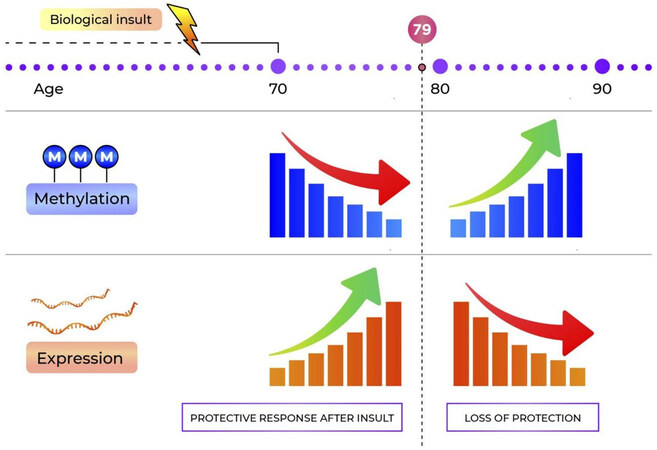
Figure 2. Model of the fluctuation in PM20D1 methylation and expression in the years leading up to a diagnosis of Alzheimer’s disorder.
The role of methylation patterns in PM20D1, both in the absence of AD symptoms and throughout disease progression, merits further investigation. The triggers involved in the switch from hypo- to hypermethylation have not been elucidated. For example, it has been suggested that changes in the expression levels of epigenetic regulator MeCP2 could play a role in PM20D1 repression in AD[46]. Importantly, there is evidence of a strong correlation between blood and brain methylation levels in the gene[7,8], which will greatly facilitate the study of gene expression in larger populations.
OBESITY
PM20D1 has been pointed out as a strong candidate to combat obesity by inducing UCP1-independent adaptive thermogenesis. It was previously postulated that, similar to other metalloproteinases, it could be associated with obesity in rodent models[47]. Several lines of evidence provide support in favor of this hypothesis. By increasing the levels of the enzyme PM20D1 in the blood of mice, their respiration as well as the concentration of NAAs are increased. In addition, through a direct supply of NAAs, an improvement in glucose homeostasis and an increase in mitochondrial energy expenditure are observed[9]. Additionally, it has been reported that obese mice treated with PM20D1 on a high-fat diet had significantly less body weight gain (9%-10%) after 40 days. The weight difference was exclusively due to a 30% reduction in lean mass, and an increase in O2 and CO2 volumes was also observed[48]. In light of these results, PM20D1 or its products have been proposed as potential therapeutic agents against obesity[49].
Mouse PM20D1 shares 71% sequence identity and 86% similarity with that of humans[9]. In 2019, two peroxisome proliferator-activated receptor γ (PPARγ) binding sites were identified near the PM20D1 gene transcription start site in human adipocytes but not in mice. PPARγ activation of PM20D1 in adipocytes differs between individuals due to a single genetic difference at rs6667995, where the alternative allele (C) disrupts a PPARγ binding motif[36].
Studies in humans have generated results that seem to be in contrast to what has been observed in mice. A recent human study found a significant increase in the serum concentration of PM20D1 and two different NAAs (C18:1-Leu and C18:1-Phe) in individuals presenting overweight or obesity. Serum concentration was positively correlated with body weight, BMI, waist circumference, and waist-hip ratio, as well as parameters related to glucose dysregulation and insulin resistance. Adjusting for age and BMI, the data suggest an association with the development of insulin resistance and glucose dysregulation. A significant increase in serum PM20D1 concentration was also observed with the presence of an increasing number of metabolic syndrome components[50]. Several different explanations have been proposed for the increased PM20D1 and NAA levels in obesity: (1) it is a result of an increase in adipose tissue, because adipocytes are one of the major expression sites for PM20D1; (2) it is a defense mechanism to prevent progression of obesity through UCP1-independent thermogenesis (which would be in line with the overexpression in early cognitive impairment seen for AD); and (3) it reflects PM20D1 or NAA resistance in the body[50].
Several SNPs in the haplotype associated with PM20D1 expression levels show significant genome-wide association with BMI[36,51], while weaker associations with type 2 diabetes and HDL cholesterol levels have been reported as well[36]. The direction of these associations is consistent with expectations: variants associated with lower BMI are also associated with lower diabetes risk and higher HDL cholesterol. For the SNPs in the haplotype, the strength of association with PM20D1 expression correlates very strongly with the strength of association with BMI/obesity. However, and again, in contrast to what would be expected from results in mice studies, the haplotype associated with absent PM20D1 expression is the one associated with lower BMI[36]. Consistent with this result, hypomethylation (and presumably increased expression) of PM20D1 has been reported in a group of women with obesity compared to controls[52]. There is also one report of hypermethylation in individuals with obesity[47].
The evidence from GWAS suggests that levels of PM20D1 expression (which are at least in part genetically determined) are associated with BMI and the risk of obesity. However, independent studies have found that NAA levels do not change significantly in knockout mice for PM20D1 or humans homozygous for the haplotype associated with silenced PM20D1 expression[10,36]. This suggests that other enzymes generate and regulate levels of NAAs, and that PM20D1’s effect on the body weight phenotype could involve a mechanism different from NAA regulation.
PARKINSON’S
The genes PM20D1, SLC41A1, RAB29 (also called RAB7L1), NUCKS1, and SLC5A3 are part of the PARK16 locus[53]. Several investigations have found an association between SNPs of the PARK16 locus and idiopathic Parkinson’s disease (PD)[37,53-58]. Nevertheless, results have differed across studies and populations, with important variations in the SNPs significantly associated with PD and their allelic frequencies in the groups of cases and controls. The underlying mechanism and the PARK16 genes involved in the onset of PD are unclear, but putative mechanisms have been suggested for SLC41A1, NUCKS, and RAB29[59-62], including epistasis and allelic heterogeneity models[63,64].
Despite the lack of known mechanisms involving PM20D1 in the pathogenesis of PD, it is well known that mitochondrial dysfunction and oxidative stress are present in many PD patients[65,66]. Mitochondrial dysfunction in PD patients is evidenced by mitochondrial complex I (MCI) deficiency in substantia nigra[67,68]; this region contains the dopaminergic neurons that are lost in PD. González-Rodríguez et al. (2021) recently showed that MCI dysfunction is enough to cause progressive, human-like parkinsonism in mice[69]. The partial inhibition of MCI by drugs increases ROS production and promotes cellular oxidative stress[70,71]. These processes participate in the damage of the dopaminergic neurons in PD[72]. In addition, the dopaminergic neurons may be under increased oxidative stress, since the oxidative metabolism of dopamine produces ROS[73,74]. Given the function of PM20D1 and the role of NAAs as endogenous mitochondrial uncouplers, as well as their potential role against oxidative activity[5,75,76], it cannot be ruled out that PM20D1 has a neuroprotective effect on the development of PD.
To the best of our knowledge, only two variants in PM20D1 with significant associations have been reported. Intronic variant rs11240572 has been significantly[53,77-79] or close to significantly[80] associated with the PD phenotype; the minor allele (A) has been proposed to have a protective effect due to its higher frequency in controls than cases. In contrast, Deng et al. (2019) reported that Chinese PD patients with rs11240572-A presented a faster progression and greater deterioration of motor function than non-carriers[81]. The common coding variant rs1891460-C (p.Ile149Val) has been reported as nominally associated with a reduced PD risk in individuals of European and Ashkenazi Jewish ancestry[57]; there are no other studies that confirm this association. Few studies have sequenced PM20D1 looking for variants in PD patients[57,63], while introns and UTR regions have not been deeply explored. Therefore, the existence of unknown variants involved in PD cannot be ruled out.
There is one report of hypermethylation of PM20D1 in PD[82]. Additionally, several studies have focused on genetic variants that correlate with the expression level of the gene. The rs708723-T variant in RAB29 has been reported to be associated with Parkinson’s disease and correlates with increased methylation of CpG sites in PM20D1 in frontal cortex and cerebellar tissues, but also with the expression of RAB29 and NUCKS1[56]. In the same tissues, the risk PD variant rs823118-T, located between RAB29 and NUCKS1, increases PM20D1 methylation and affects the expression of RAB29 and NUCKS1[64]. Goldstein et al. (2021) found evidence suggesting that the risk non-coding variant rs823114-A, located upstream of the NUCKS1 gene, has an eQTL and mQTL effect on PM20D1 and other genes in the PARK16 locus[83]. Additionally, Cibulka et al. (2022) reported that minor allele A for rs708727 in SLC41A1 is associated with PD in the Slovak population[37]. As mentioned in previous sections, the Alzheimer-associated rs708727 is an mQTL and eQTL for PM20D1, and the A allele is associated with higher methylation of the PM20D1 promoter and absent expression[5-8] [Figure 1]. In addition, dementia is a common diagnosis in patients with PD, manifesting mainly in late stages of the disease[84,85]. This suggests that there may be common mechanisms in the development of PD and AD, including mechanisms of epigenetic regulation[37].
OTHER PHENOTYPES
As evidenced in the previous sections, PM20D1 is closely related to AD, obesity, and PD; however, PM20D1 has been associated with a wide variety of other phenotypes. Among those is asthma, where the hypomethylation of probe cg14893161 was initially shown in babies born to mothers with asthma and atopic mothers without asthma[86]. A direct association between hypermethylation of PM20D1 and respiratory allergy, specifically for probe cg11965913, has also been identified; expression of PM20D1 is regulated by the TLR5 pathway, which is closely related to allergic responses[87]. In addition to a respiratory allergic response, hypomethylation in the gene has also been associated with food allergies[88].
Changes in the methylation levels of PM20D1 have been reported in several additional phenotypes. In the case of psoriasis, an upregulation in gene methylation has been reported in affected individuals[89]. A differentially methylated region at PM20D1 has also been identified for multiple sclerosis[90].
PM20D1 is also susceptible to epigenetic changes as a result of external factors, such as lifetime events and environmental conditions. In individuals who have suffered child abuse, there is evidence of hypermethylation of PM20D1[91]. PM20D1 has also been involved in the response mechanism of cold exposure, in which there is an upregulation of PM20D1 under conditions of constant exposure to cold[92].
There is increasing evidence that PM20D1 plays a role in several metabolic conditions, such as obesity and diabetes (discussed above). Additionally, a suggestive pleiotropic association with polycystic ovary syndrome has been described for the gene; this is a metabolic condition closely related with obesity and diabetes[93]. For familial hypercholesterolemia, DMRs have been found within PM20D1, specifically in the cg14893161 probe[94]. On the other hand, decreased serum PM20D1 has been associated with severity in carotid atherosclerosis patients[95].
Furthermore, the very diverse phenotypes related to PM20D1 include stroke, a condition that was determined to present hypermethylation within PM20D1 independent of the body mass index[96]. A study of breast tissue from healthy women found differences in PM20D1 methylation according to ethnic origin[97]. Additionally, in an epigenome-wide association study for COVID-19 severity, the PM20D1 gene was found to be differentially methylated[98].
PM20D1 has a potential role in cancer as well; differentially methylated regions at PM20D1 have been reported for different types of cancer. As a first example, the methylation level of PM20D1 (together with other genes) has been proposed to allow detection of the presence of lung cancer, as well as to characterize the type of tumor[99]. Additionally, hypermethylation at PM20D1 sites has been found in hepatocellular carcinoma and acute myeloid leukemia, suggesting that the gene is downregulated or totally silenced[100,101].
Finally, it is important to highlight the role of PM20D1 in neurological disorders. In addition to its involvement in AD and PD (as detailed in previous sections), hypermethylation of CpG sites in PM20D1 (and presumably reduced expression) has been associated with an increased risk of suffering chronic postsurgical pain[102]. Additionally, it has been reported that PM20D1 is differentially methylated between different types of epilepsy (focal vs. generalized epilepsy)[103].
In most cases, the evidence supporting the role of PM20D1 in these disorders comes from a single report with a small sample size. Therefore, further studies are needed to confirm these associations, as well as to understand the mechanisms involved.
CONCLUSION
Evidence supporting the role of the PM20D1 gene in several heterogeneous disorders has been steadily accumulating in recent years. Thus far, it appears that the main effect of this gene on phenotype comes from changes in its expression levels, rather than from genetic variants that affect its structure or function. However, genetic variants in the genomic region close to PM20D1 do play a role in the regulation of the gene’s expression levels, as has been shown by the multiple studies confirming the existence of these mQTLs and eQTLs. This stresses the importance of performing comprehensive studies that explore genetic variation, methylation, and expression at the same time. Although the direction of change in methylation or expression of PM20D1 varies among disorders, many studies report hypermethylation or reduced expression of PM20D1 (or a higher frequency of the reduced expression-associated haplotype) in affected individuals. This (and other functional evidence) supports the idea of the protective role of PM20D1, which is lost in affected individuals with silenced expression. PM20D1 has been shown to activate mitochondrial uncoupling, which plays a role in response to oxidative stress. Oxidative stress is known to be involved in the development and progression of several PM20D1-associated disorders, including obesity, Alzheimer’s disease, and Parkinson’s disease, and could potentially be the common link among them. The exact biological mechanisms involved in each case await elucidation, which could potentially open up promising avenues for treatment.
DECLARATIONS
AcknowledgmentsWe thank Gabriel Jiménez-Huezo for creating Figures 1 and 2.
Authors’ contributionsContributed to the conception of the study: Raventós-Vorst H, Chavarría-Soley G
Drafting of the manuscript: Garro-Núñez D, Mora-Cubillo P, Fonseca-Bone S, Picado-Martínez MJ, Fonseca-Brenes M, Chavarría-Soley G
Supervision: Garro-Núñez D, Raventós-Vorst H, Chavarría-Soley G
Availability of data and materialsNot applicable.
Financial Support and SponsorshipNot applicable.
Conflicts of InterestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for PublicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Visscher PM, Wray NR, Zhang Q, et al. 10 Years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017;101:5-22.
2. Li S, Tollefsbol TO. DNA methylation methods: global DNA methylation and methylomic analyses. Methods 2021;187:28-43.
4. Kim JT, Li VL, Terrell SM, Fischer CR, Long JZ. Family-wide annotation of enzymatic pathways by parallel in vivo metabolomics. Cell Chem Biol 2019;26:1623-1629.e3.
5. Sanchez-Mut JV, Glauser L, Monk D, Gräff J. Comprehensive analysis of PM20D1 QTL in Alzheimer’s disease. Clin Epigenetics 2020;12:20.
6. Sanchez-Mut JV, Heyn H, Silva BA, et al. PM20D1 is a quantitative trait locus associated with Alzheimer’s disease. Nat Med 2018;24:598-603.
7. Wang Q, Chen Y, Readhead B, et al. Longitudinal data in peripheral blood confirm that PM20D1 is a quantitative trait locus (QTL) for Alzheimer’s disease and implicate its dynamic role in disease progression. Clin Epigenetics 2020;12:189.
8. Coto-Vílchez C, Martínez-Magaña JJ, Mora-Villalobos L, et al. Genome-wide DNA methylation profiling in nonagenarians suggests an effect of PM20D1 in late onset Alzheimer’s disease. CNS Spectr 2021:1-27.
9. Long JZ, Svensson KJ, Bateman LA, et al. The secreted enzyme PM20D1 regulates lipidated amino acid uncouplers of mitochondria. Cell 2016;166:424-35.
10. Long JZ, Roche AM, Berdan CA, et al. Ablation of PM20D1 reveals N-acyl amino acid control of metabolism and nociception. Proc Natl Acad Sci USA 2018;115:E6937-45.
11. Schaum N, Karkanias J, Neff NF, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367-72.
12. GTEx portal. Available from: https://gtexportal.org/home/gene/PM20D1 [Last accessed on 12 August 2022].
13. HBT - Human Brain Transcriptome. Available from: https://hbatlas.org/ [Last accessed on 12 August 2022].
14. Brain RNA-Seq. Available from: http://brainrnaseq.org/ [Last accessed on 12 August 2022].
15. Velmeshev D, Schirmer L, Jung D, et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019;364:685-9.
16. Kim JT, Jedrychowski MP, Wei W, et al. A Plasma protein network regulates PM20D1 and N-Acyl amino acid bioactivity. Cell Chem Biol 2020;27:1130-1139.e4.
17. Hanuš L, Shohami E, Bab I, Mechoulam R. N-Acyl amino acids and their impact on biological processes. Biofactors 2014;40:381-8.
18. Milman G, Maor Y, Abu-Lafi S, et al. N-arachidonoyl L-serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc Natl Acad Sci USA 2006;103:2428-33.
19. Tosini G, Ye K, Iuvone PM. N-acetylserotonin: neuroprotection, neurogenesis, and the sleepy brain. Neuroscientist 2012;18:645-53.
20. Rimmerman N, Bradshaw HB, Hughes HV, et al. N-palmitoyl glycine, a novel endogenous lipid that acts as a modulator of calcium influx and nitric oxide production in sensory neurons. Mol Pharmacol 2008;74:213-24.
21. Huang SM, Bisogno T, Petros TJ, et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J Biol Chem 2001;276:42639-44.
22. Tan B, O’Dell DK, Yu YW, et al. Identification of endogenous acyl amino acids based on a targeted lipidomics approach. J Lipid Res 2010;51:112-9.
23. Lin H, Long JZ, Roche AM, et al. Discovery of hydrolysis-resistant isoindoline N-Acyl amino acid analogues that stimulate mitochondrial respiration. J Med Chem 2018;61:3224-30.
24. Keipert S, Kutschke M, Ost M, et al. Long-term cold adaptation does not require FGF21 or UCP1. Cell Metab 2017;26:437-446.e5.
25. Gao Y, Shabalina IG, Braz GRF, Cannon B, Yang G, Nedergaard J. Establishing the potency of N-acyl amino acids versus conventional fatty acids as thermogenic uncouplers in cells and mitochondria from different tissues. Biochim Biophys Acta Bioenerg 2022;1863:148542.
26. Mookerjee SA, Divakaruni AS, Jastroch M, Brand MD. Mitochondrial uncoupling and lifespan. Mech Ageing Dev 2010;131:463-72.
27. Brand M. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exper Gerontol 2000;35:811-20.
28. Zaninovich ÁA. Role of the uncoupling proteins UCP1, UCP2 and UCP3 in energy balance, type 2 diabetes and obesity: synergism with the thyroid. Med B Aires 2005;65:163-9.
29. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 2005;39:359-407.
30. Papa S, Skulachev VP. Reactive oxygen species, mitochondria, apoptosis and aging. In: Gellerich FN, Zierz S, editors. Detection of mitochondrial diseases. Boston: Springer; 1997. pp. 305-19.
31. Brand MD, Affourtit C, Esteves TC, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 2004;37:755-67.
32. Kokoszka JE, Waymire KG, Levy SE, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004;427:461-5.
33. Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol 2018;15:490-503.
34. consortium. human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648-60.
36. Benson KK, Hu W, Weller AH, et al. Natural human genetic variation determines basal and inducible expression of PM20D1, an obesity-associated gene. Proc Natl Acad Sci USA 2019;116:23232-42.
37. Cibulka M, Brodnanova M, Grendar M, et al. Alzheimer’s disease-associated SNP rs708727 in SLC41A1 may increase risk for parkinson’s disease: report from enlarged slovak study. Int J Mol Sci 2022;23:1604.
38. Greenawalt DM, Dobrin R, Chudin E, et al. A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res 2011;21:1008-16.
39. Gunasekara CJ, Scott CA, Laritsky E, et al. A genomic atlas of systemic interindividual epigenetic variation in humans. Genome Biol 2019;20:105.
40. Nica AC, Parts L, Glass D, et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 2011;7:e1002003.
41. Civelek M, Wu Y, Pan C, et al. Genetic regulation of adipose gene expression and cardio-metabolic traits. Am J Hum Genet 2017;100:428-43.
42. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434-43.
43. Heyn H, Moran S, Hernando-Herraez I, et al. DNA methylation contributes to natural human variation. Genome Res 2013;23:1363-72.
44. Li QS, Vasanthakumar A, Davis JW, et al. Association of peripheral blood DNA methylation level with Alzheimer’s disease progression. Clin Epigenetics 2021;13:191.
45. Pérez RF, Alba-Linares JJ, Tejedor JR, et al. Blood DNA methylation patterns in older adults with evolving dementia. J Gerontol A Biol Sci Med Sci 2022:glac068.
46. Kim B, Choi Y, Kim HS, Im HI. Methyl-CpG binding protein 2 in alzheimer dementia. Int Neurourol J 2019;23:S72-81.
47. Feinberg AP, Irizarry RA, Fradin D, et al. Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med 2010;2:49ra67.
48. Larrick JW, Larrick JW, Mendelsohn AR. Uncoupling mitochondrial respiration for diabesity. Rejuvenation Res 2016;19:337-40.
50. Yang R, Hu Y, Lee CH, et al. PM20D1 is a circulating biomarker closely associated with obesity, insulin resistance and metabolic syndrome. Eur J Endocrinol 2021;186:151-61.
51. Yengo L, Sidorenko J, Kemper KE, et al. Meta-analysis of genome-wide association studies for height and body mass index in ~700000 individuals of European ancestry. Hum Mol Genet 2018;27:3641-9.
52. Noronha NY, Barato M, Sae-lee C, et al. Novel Zinc-related differentially methylated regions in leukocytes of women with and without obesity. Front Nutr 2022;9:785281.
53. Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 2009;41:1303-7.
54. Yan Y, Tian J, Mo X, et al. Genetic variants in the RAB7L1 and SLC41A1 genes of the PARK16 locus in chinese parkinson’s disease patients. Int J Neurosci 2011;121:632-6.
55. Tucci A, Nalls MA, Houlden H, et al. Genetic variability at the PARK16 locus. Eur J Hum Genet 2010;18:1356-9.
56. Parkinson’s Disease Genomics Consortium (IPDGC); Wellcome Trust Case Control Consortium 2 (WTCCC2). A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet 2011;7:e1002142.
57. Rudakou U, Yu E, Krohn L, et al. Targeted sequencing of Parkinson’s disease loci genes highlights SYT11, FGF20 and other associations. Brain 2021;144:462-72.
58. Gan-Or Z, Bar-Shira A, Dahary D, et al. Association of sequence alterations in the putative promoter of RAB7L1 with a reduced parkinson disease risk. Arch Neurol 2012;69:105-10.
59. Kolisek M, Sponder G, Mastrototaro L, et al. Substitution p.A350V in Na+/Mg2+ exchanger SLC41A1, potentially associated with Parkinson’s disease, is a gain-of-function mutation. PLoS One 2013;8:e71096.
60. Lin CH, Wu YR, Chen WL, et al. Variant R244H in Na+/Mg2+ exchanger SLC41A1 in Taiwanese Parkinson’s disease is associated with loss of Mg2+ efflux function. Parkinsonism Relat Disord 2014;20:600-3.
61. Bai Y, Dong L, Huang X, Zheng S, Qiu P, Lan F. Associations of rs823128, rs1572931, and rs823156 polymorphisms with reduced Parkinson’s disease risks. Neuroreport 2017;28:936-41.
62. Singh S, Seth PK. Functional association between NUCKS1 gene and Parkinson disease: A potential susceptibility biomarker. Bioinformation 2019;15:548-56.
63. Pihlstrøm L, Rengmark A, Bjørnarå KA, et al. Fine mapping and resequencing of the PARK16 locus in Parkinson’s disease. J Hum Genet 2015;60:357-62.
64. Nalls MA, Pankratz N, Lill CM, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 2014;46:989-93.
65. Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol Dis 2013;51:35-42.
66. Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol 2013;106-107:17-32.
67. Mann VM, Cooper JM, Daniel SE, et al. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann Neurol 1994;36:876-81.
68. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 1990;54:823-7.
69. González-Rodríguez P, Zampese E, Stout KA, et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021;599:650-6.
70. Votyakova TV, Reynolds IJ. Ca2+-induced permeabilization promotes free radical release from rat brain mitochondria with partially inhibited complex I. J Neurochem 2005;93:526-37.
71. Koopman WJ, Verkaart S, Visch HJ, et al. Inhibition of complex I of the electron transport chain causes O2-mediated mitochondrial outgrowth. Am J Physiol Cell Physiol 2005;288:C1440-50.
73. Hastings TG. Enzymatic oxidation of dopamine: the role of prostaglandin H synthase. J Neurochem 1995;64:919-24.
74. Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J Neurochem 1999;73:1127-37.
75. Buc-Calderon P, Roberfroid M. Increase in the survival time of mice exposed to ionizing radiation by a new class of free radical scavengers. Experientia 1990;46:708-10.
76. Suzen S, Gurkok G, Coban T. Novel N-acyl dehydroalanine derivatives as antioxidants: studies on rat liver lipid peroxidation levels and DPPH free radical scavenging activity. J Enzyme Inhib Med Chem 2006;21:179-85.
77. Chang KH, Chen CM, Chen YC, et al. Association between PARK16 and Parkinson’s disease in the Han Chinese population: a meta-analysis. Neurobiol Aging 2013;34:2442.e5-9.
78. Tan EK, Kwok HH, Tan LC, et al. Analysis of GWAS-linked loci in Parkinson disease reaffirms PARK16 as a susceptibility locus. Neurology 2010;75:508-12.
79. Yan YP, Mo XY, Tian J, et al. An association between the PARK16 locus and Parkinson’s disease in a cohort from eastern China. Parkinsonism Relat Disord 2011;17:737-9.
80. Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009;41:1308-12.
81. Deng X, Xiao B, Allen JC, et al. Parkinson’s disease GWAS-linked Park16 carriers show greater motor progression. J Med Genet 2019;56:765-8.
82. Henderson AR, Wang Q, Meechoovet B, et al. DNA methylation and expression profiles of whole blood in parkinson’s disease. Front Genet 2021;12:640266.
83. Goldstein O, Gana-Weisz M, Casey F, et al. PARK16 locus: differential effects of the non-coding rs823114 on Parkinson’s disease risk, RNA expression, and DNA methylation. J Genet Genomics 2021;48:341-5.
84. Meireles J, Massano J. Cognitive impairment and dementia in Parkinson’s disease: clinical features, diagnosis, and management. Front Neurol 2012;3:88.
85. Hanagasi HA, Tufekcioglu Z, Emre M. Dementia in Parkinson’s disease. J Neurol Sci 2017;374:26-31.
86. Gunawardhana LP, Baines KJ, Mattes J, Murphy VE, Simpson JL, Gibson PG. Differential DNA methylation profiles of infants exposed to maternal asthma during pregnancy. Pediatr Pulmonol 2014;49:852-62.
87. Langie SAS, Szarc vel Szic K, Declerck K, et al. Whole-genome saliva and blood DNA methylation profiling in individuals with a respiratory allergy. PLoS ONE 2016;11:e0151109.
88. Imran S, Neeland MR, Koplin J, et al. Epigenetic programming underpins B-cell dysfunction in peanut and multi-food allergy. Clin Transl Immunol 2021;10:e1324.
89. Li X, Zhao X, Xing J, et al. Different epigenome regulation and transcriptome expression of CD4+ and CD8+ T cells from monozygotic twins discordant for psoriasis. Australas J Dermatol 2020;61:e388-94.
90. Maltby VE, Lea RA, Sanders KA, et al. Differential methylation at MHC in CD4+ T cells is associated with multiple sclerosis independently of HLA-DRB1. MHC 2017;9:71.
91. Suderman M, Borghol N, Pappas JJ, et al. Childhood abuse is associated with methylation of multiple loci in adult DNA. BMC Med Genomics 2014;7:13.
92. Gao Y, Qimuge NR, Qin J, et al. Acute and chronic cold exposure differentially affects the browning of porcine white adipose tissue. Animal 2018;12:1435-41.
93. Sun Q, Gao Y, Yang J, Lu J, Feng W, Yang W. Mendelian randomization analysis identified potential genes pleiotropically associated with polycystic ovary syndrome. Reprod Sci 2022;29:1028-37.
94. Guay SP, Brisson D, Mathieu P, Bossé Y, Gaudet D, Bouchard L. A study in familial hypercholesterolemia suggests reduced methylomic plasticity in men with coronary artery disease. Epigenomics 2015;7:17-34.
95. Huang X, He P, Wu L. Clinical significance of peptidase M20 domain containing 1 ii patients with carotid atherosclerosis. Arq Bras Cardiol ;2022:S0066-782X2022005005206.
96. Gómez-Úriz AM, Milagro FI, Mansego ML, et al. Obesity and ischemic stroke modulate the methylation levels of KCNQ1 in white blood cells. Hum Mol Genet 2015;24:1432-40.
97. Song MA, Brasky TM, Marian C, et al. Racial differences in genome-wide methylation profiling and gene expression in breast tissues from healthy women. Epigenetics 2015;10:1177-87.
98. Castro de Moura M, Davalos V, Planas-Serra L, et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 2021;66:103339.
99. Goldmann T, Schmitt B, Müller J, et al. DNA methylation profiles of bronchoscopic biopsies for the diagnosis of lung cancer. Clin Epigenetics 2021;13:38.
100. Revill K, Wang T, Lachenmayer A, et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology 2013;145:1424-35.e1.
101. Huang J, Liu Z, Sun Y, et al. Use of methylation profiling to identify significant differentially methylated genes in bone marrow mesenchymal stromal cells from acute myeloid leukemia. Int J Mol Med 2018;41:679-86.
102. Chidambaran V, Zhang X, Pilipenko V, et al. Methylation quantitative trait locus analysis of chronic postsurgical pain uncovers epigenetic mediators of genetic risk. Epigenomics 2021;13:613-30.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Garro-Núñez D, Mora-Cubillo P, Fonseca-Bone S, Picado-Martínez MJ, Fonseca-Brenes M, Raventós-Vorst H, Chavarría-Soley G. The many roles of the Alzheimer-associated gene PM20D1. J Transl Genet Genom 2022;6:361-74. http://dx.doi.org/10.20517/jtgg.2022.10
AMA Style
Garro-Núñez D, Mora-Cubillo P, Fonseca-Bone S, Picado-Martínez MJ, Fonseca-Brenes M, Raventós-Vorst H, Chavarría-Soley G. The many roles of the Alzheimer-associated gene PM20D1. Journal of Translational Genetics and Genomics. 2022; 6(3): 361-74. http://dx.doi.org/10.20517/jtgg.2022.10
Chicago/Turabian Style
Garro-Núñez, Diana, Pablo Mora-Cubillo, Sammy Fonseca-Bone, María Jesús Picado-Martínez, Michael Fonseca-Brenes, Henriette Raventós-Vorst, Gabriela Chavarría-Soley. 2022. "The many roles of the Alzheimer-associated gene PM20D1" Journal of Translational Genetics and Genomics. 6, no.3: 361-74. http://dx.doi.org/10.20517/jtgg.2022.10
ACS Style
Garro-Núñez, D.; Mora-Cubillo P.; Fonseca-Bone S.; Picado-Martínez MJ.; Fonseca-Brenes M.; Raventós-Vorst H.; Chavarría-Soley G. The many roles of the Alzheimer-associated gene PM20D1. J. Transl. Genet. Genom. 2022, 6, 361-74. http://dx.doi.org/10.20517/jtgg.2022.10
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 20 clicks
Cite This Article 20 clicks

 Like This Article 2
likes
Like This Article 2
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.