
Genetic risk and its role in primary prevention of CAD

Abstract
Coronary artery disease (CAD) is a pandemic disease and the number one cause of death in the world. Predisposition to CAD is about 50% acquired and 50% genetic. CAD prevention has been proven in randomized clinical trials with statin therapy. However, primary prevention is limited by the lack of biomarkers to detect asymptomatic young individuals at risk. Traditional risk factors (TRFs) such as hypertension or Type 2 Diabetes are age-dependent and often not present until the sixth or seventh decade. In contrast, genetic risk determined at conception is potentially a biomarker to detect young individuals at risk for CAD. The first genetic risk variant for CAD (9p21) was discovered in 2007, and subsequently, over 200 risk variants for CAD were discovered. A genetic risk score (GRS) based on the genetic risk variants for CAD was evaluated in over one million individuals. Retrospective analysis of clinical trials assessing the effect of statin therapy showed that individuals with the highest GRS had the highest risk for cardiac events and also the most benefit from lowering cholesterol. In a recent study of 55,685 individuals, those with the highest GRS (20%) had a 91% higher risk for cardiac events. Furthermore, those with high genetic risk on a favorable lifestyle had 46% fewer cardiac events than those with an unfavorable lifestyle. The GRS is superior and independent of TRFs. Incorporation into clinical practice will be a paradigm shift in preventing this pandemic.
Keywords
INTRODUCTION
Coronary Artery Disease (CAD) has, for decades, been the number one cause of death in the Western World[1]. Furthermore, CAD has now replaced infection as the first cause of death in middle- and low-income countries[1]. Despite this pandemic spread, the Western World has made remarkable advances in both the prevention and treatment of CAD. In the past 30 years, the number of cases of myocardial infarction in the United States has gone from over 900,000 to less than 500,000 per year, a reduction of about 50%[2]. This reduction has been mediated through primary and secondary prevention as well as significant improvement in treatment modalities. Is it possible from the lessons learned in the Western World to initiate early primary prevention in high-, middle-, and low-income countries?
This is perhaps a more pivotal time for both global and comprehensive prevention of CAD. Preventive measures have had tremendous success; however, they have been less comprehensive than desired. Epidemiologists have recognized for several decades that predisposition for CAD is equally divided between risk factors due to genetic predisposition and the risk associated with lifestyle and environmental risk factors[3]. The discoveries over the last 15 years of genetic risk variants predisposing to CAD enable one to determine the risk early in life independent of age and acquired factors[3,4]. Secondly, it is estimated by the World Health Organization (WHO) that over 60% of the morbidity and mortality from CAD occurs in low and middle-income countries[1]. This review will summarize the discovery of genetic risk variants for CAD, the development of a genetic risk score for CAD and its potential to risk stratify for primary prevention of CAD [Figure 1].
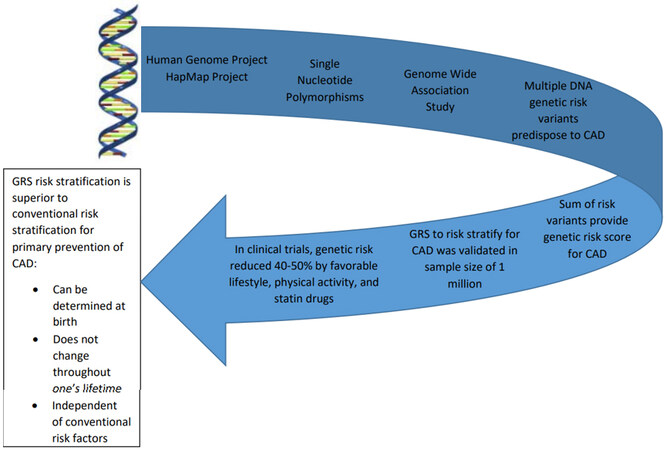
Figure 1. Illustration of the pathway from genetic discovery to clinical application. The Human Genome Project and the HapMap Project made available SNPs that could be used as markers to span the human genome (leading to Genome-Wide Association Studies). Multiple genetic risk variants were discovered and the total risk was summarized in a single number referred to as the Genetic Risk Score (GRS). The GRS was evaluated as a means to risk-stratify for CAD in several studies with an overall sample size of over one million. Genetic risk for CAD was reduced by 40%-50% due to lifestyle changes and drug therapy. Genetic risk is determined at conception and does not change in one’s lifetime. GRS is superior because it enables risk stratification for CAD at an early age prior to the development of conventional risk factors in males or females.
CHOLESTEROL PIVOTAL TO THE ETIOLOGY AND PATHOGENESIS OF CAD
The development of atherosclerosis in the vasculature of any organ occurs early in life with the tendency to slowly and relentlessly progress. Autopsy studies performed on young individuals, such as during the Korean War[4], indicate the presence of early coronary atherosclerosis even in the teenage years. This process remains subclinical until it reaches a certain threshold which induces clinical manifestations such as sudden cardiac death and myocardial infarction. This threshold is usually associated with a 30% to 40% reduction in the diameter of the coronary vessel. Despite the adequate coronary flow adequate, abnormalities in the vessel wall predispose to the formation of thrombosis, which can completely occlude the vessel precipitating myocardial infarction and its clinical sequelae. In males, the peak incidence of heart attacks occurs in the sixth decade and in females in the seventh decade. The later onset of clinical manifestations in the female is due to hormonal protection which slows the progress of coronary atherosclerosis until it is lost with the onset of menopause. Multiple clinical and experimental studies strongly indicate that plasma LDL-C is the main culprit inducing atherosclerosis. The genetic, epidemiological, and clinical research is well summarized in a consensus document published by the European Society of Cardiology summarizing over 200 studies involving over two million participants experiencing more than 150,000 cardiovascular events[5]. The authors elucidate a dose-dependent log-linear association between the concentration of plasma LDL-C and the incidence of CAD. A cohort recently studied from the Framingham Offspring concluded that the risk of CAD from plasma LDL-C, in addition to the intensity of the LDL-C concentration, is also related to the duration of exposure to LDL-C, such that the risk of CAD doubles for each additional decade of exposure[6]. There is also current evidence from the discovery of genetic risk variants predisposing to CAD and the recent randomized placebo-controlled clinical trial CANTOS[7] that inflammation also plays a role in the pathogenesis of CAD and its clinical manifestations.
CAD HAS BEEN PROVEN TO BE PREVENTABLE
CAD is a pandemic disease that has increased rapidly in low- and mid-income countries as they have adopted more and more of the western diet and lifestyles. CAD has been studied extensively since the 1950s and much has been learned about its prevention and management. In the western world, the identification of conventional risk factors for CAD, commonly referred to as traditional risk factors (TRFs) including age, cholesterol, smoking, diabetes, obesity, hypertension, and family history, has enabled the application of primary and secondary prevention. Multiple studies have documented that decreasing the risk associated with these factors has had a major impact on the western world[8,9]. In the United States, despite CAD remaining the most common cause of death, cardiac mortality has decreased by 50% in the past 30 years due to a combination of prevention and improved management[2]. Secondary prevention of CAD has been accepted widely and the most successful, while primary prevention is expected to be even more fruitful.
Lifestyle changes such as smoking cessation is very effective in reducing the risk for CAD and drug therapy can reduce the plasma LDL-C. The drugs are primarily statins since they were the only class of drugs until recently shown to be safe and efficacious for reducing cholesterol. The first placebo-controlled clinical trial published in 1994 confirmed that a reduction in plasma LDL-C by simvastatin was associated with a significant reduction in cardiac morbidity and mortality[10]. Afterward, multiple clinical trials are performed, all of which consistently confirm that lowering plasma LDL-C by a variety of statins is associated with a 30%-40% reduction in cardiac morbidity and mortality. Additionally, all of the statins administered in these trials are also consistently shown to be of high safety. Mainly skeletal muscular side effects are in the range of 1%-2% and are reversible. There is also evidence that statins may increase one’s predisposition to diabetes and cataracts. The safety and efficacy of statin therapy in the prevention of CAD are well summarized in a meta-analysis by the Cholesterol Treatment Trialist Analysis of more than 170,000 individuals[9]. Many of the statins proven to be safe and efficacious are now off-patent and available through generic providers enabling them to be inexpensive and available for management around the world. Statin therapy is no longer a therapy available only to high-income but also accessible and financially feasible for middle- and low-income countries.
The drug armamentarium to decrease plasma LDL-C has been significantly enriched in the past decade. Randomized placebo-controlled clinical trials utilizing PCSK9 inhibitors have shown marked reductions in cardiac morbidity and mortality[11-14]. Furthermore, their mechanism of action is distinct from that of statins and thus complementary. Statins inhibit the rate-limiting enzyme responsible for the synthesis of cholesterol, while PCSK9 inhibition prevents the breakdown of the LDL-C receptor. Inhibiting the degradation of the LDL-C receptor, the main mechanism whereby the liver removes LDL-C from the blood, leads to faster and more extensive removal of plasma LDL-C.
The recent discovery of Angiopoietin-like 3[15,16] added yet another potential class of drugs with a distinctly different mechanism to lower plasma cholesterol. Evinacumab was used as an antibody to inhibit Angiopoietin-like 3 and was shown to reduce plasma LDL-C by 41%[17]. This drug inhibits endothelial lipase, which leads to decreased plasma triglycerides and other lipids[18,19].
GENETIC PREDISPOSITION ACCOUNTS FOR 50% OF THE RISK FOR CAD
For decades epidemiologists have estimated that genetic predisposition accounts for about 50% of the risk for CAD[3] based on studies on identical twins and family histories. Results of the Danish Twin Registry Study showed monozygotic twins compared with dizygotic twins experienced a frequency of CAD of 44% vs. 14%, respectively[20]. Studies out of Utah emphasized the importance of genetic risk to predisposition to CAD. It was observed that 14% of the population has a family history of CAD which accounts for 72% of the cases of premature CAD[21] and 48% of all CAD. Similarly, 11% of the population has a family history of stroke and 80% of strokes occur in this cohort. Studies from the town of Framingham show men with a family history of CAD had a 2.6-fold increased risk of CAD, and women a 2.3-fold increased risk[22]. Recent studies such as Interheart[23] and Procam[24] show a similar increased risk for CAD in members with a family history.
DEVELOPMENT OF A GENOME-WIDE SEARCH FOR GENETIC RISK VARIANTS
In the 90s, geneticists hypothesized that, unlike rare Mendelian disorders, CAD, like any common disorder (diabetes), is polygenic and transmitted by common variants[25-28]. Furthermore, it was postulated that these common variants would be single nucleotide polymorphisms distributed throughout the genome[28]. These disorders, different from single gene Mendelian disorders (e.g., Familial Hypertrophic Cardiomyopathy), are not due to a predominant gene and do not manifest a phenotype exhibiting a Mendelian dominant or recessive pattern. Thus, linkage analysis of pedigrees, the preferred method to pursue genes responsible for single gene disorders, would not be appropriate for polygenic disorders[27-29]. Furthermore, single gene disorders require only a few hundred DNA markers spaced at intervals of 10,000,000 bases to perform linkage analysis of pedigrees[30]. The preferred approach for polygenic disorders is the Case-Control Association Study (CCAS)[28,29], which in design may utilize the direct or the indirect approach. The CCAS compares the frequency of a potential risk sequence in individuals with the disease to that of unrelated controls. If the frequency is significantly greater in cases than in controls, it would indicate the sequence is in a region of DNA that predisposes to the disease. The direct approach referred to as the candidate gene approach selects potential candidate genes. These candidates are selected based on features such as function which are likely to be associated with increased risk for the disease. The frequency of the candidate in the cases is compared to that of controls. This, by definition, is a biased approach and limited to known candidate genes. The more appropriate and less biased approach is that of the indirect CCAS, in which one compares the frequency of a DNA marker in cases to that of controls. If the frequency of a DNA marker in the cases is greater than that of controls, it would indicate that the marker is in a DNA region associated with increased predisposition to the disease. This approach requires no presupposition of which marker or gene is more likely to be the candidate risk gene. Sensitivity would be greatly increased if the DNA markers were analyzed for association distributed throughout the genome. Kruglyak et al. predicted one would need hundreds of thousands of DNA markers spaced at intervals of not more than 3000 bases[31].
In 1998 Wang et al. estimated there were only a few thousand single-nucleotide polymorphisms (SNPs) available, which were not enough to span the human genome, so the candidate gene approach was adopted in large part because of the lack of DNA markers to perform the indirect CCAS[29]. Multiple studies utilizing the indirect approach were performed. These results were flawed, and none of the more than 100 genes claimed to be associated with CAD were subsequently confirmed[32,33].
Several scientific contributions made possible the application of the indirect Case-Control Association approach based on markers distributed throughout the human genome referred to as a Genome-Wide Association Study (GWAS). The initial contribution was the sequencing of the human genome in 2000[34], followed by the HapMap Project in 2005[35]. The HapMap project annotated over a million DNA markers. The markers identified by HapMap were SNPs distributed throughout the genome. This made it possible to analyze SNPs as DNA markers spanning the genome. The number of SNPs in the human genome is fairly constant at about five million. These SNPs account for over 80% of the unique features of the human phenotype including predisposition to disease[36]. These contributions in 2005 coincided with the development of computerized platforms enabling one to genotype and analyze microarrays containing millions of SNPs as DNA markers[37]. A microarray with one million markers made it possible to genotype markers at intervals of 3000 bases. However, it became evident there was a statistical concern. Utilizing one million SNPs to genotype the human genome was similar to analyzing one million endpoints. If one used a P value of 0.05 for statistical significance, it would be associated with 50,000 false positives. This required a statistical correction. Risch et al. proposed the Bonferroni correction consisting of dividing one million into 0.05, giving a P value of 0.00000005, which is usually referred to as 5 × 10-8 and now is referred to as the genome-wide significant P value[25]. At the time, it was recognized that a Bonferroni correction might be overly stringent, but it provided a standardized method for statistical interpretation and comparison across studies. Additionally[38,39], it was proposed that all risk variants for CAD reaching genome-wide significance should be replicated in an independent population. The SNPs selected for the GWAS are commonly occurring SNPs and are unlikely to detect rare genetic risk variants with a population frequency of less than 5%.
DISCOVERY OF 9P21, THE FIRST GENETIC RISK VARIANT FOR CAD
The components to perform a GWAS were now available, and the challenge was to obtain adequate samples for rapid high-throughput genotyping. In 2007 we[40] in Ottawa and our collaborators in Texas, Huston and Denmark and the deCODE Group[41] in Iceland simultaneously and independently discovered 9p21 as the first genetic risk variant for CAD. The initial microarray had only 10,000 SNPs, followed by an expansion to 500,000 SNPs. The Ottawa Heart Study-1 recruited cases with premature CAD (< 60 years). To increase the sensitivity, we did sequential screening with case-control comparisons at a minimal significance threshold of < 0.02. Initial analysis of the genotypes identified 50 SNPs significantly associated with CAD, but only two SNPs were replicated in an independent cohort of the Atherosclerosis Risk Community Study (ARIC). Both of these SNPs were situated within 20,000 bases of each other on chromosome 9p21. These SNPs were validated in three independent populations: The Copenhagen City Heart Study (CCHS), The Dallas Heart Study (DHS), and the Ottawa Heart Study-3 (OHS-3). The total number of cases and controls was 24,425. The group in Iceland, under the leadership of Helgadottir, utilized a sample size of 17,354. Their independent and simultaneous effort led to the discovery of 9p21 as a risk variant for CAD and was published in the same issue of Science. The Wellcome Trust Group shortly after that confirmed 9p21 as a risk variant for CAD in a sample size of 17,000[42]. The results of all three groups, although independent, concluded that 9p21 is a risk variant for CAD.
The features of the 9p21 risk variant further enhanced and confirmed the hypothesis that CAD was due to multiple common DNA risk variants, with each transferring only minimal risk for CAD. The 9p21 risk variant was associated with the increased relative risk for CAD of only 25% per copy. The 9p21 risk variant is estimated to occur in about 75% of the world’s population. The risk mediated by 9p21 was independent of all known conventional risk factors and was not located in a protein coding region. It signified that even larger sample sizes would be required to identify most of these risk variants. Within two years, 9p21 was confirmed throughout the world by multiple investigators in multiple populations, including Caucasians[43], Chinese[44], Koreans[45], Italians[46], Japanese[47], South East Asians in Pakistan[48], India[48], and multiple ethnic groups in South America[49]. A study on African Americans, while limited in sample size, suggested 9p21 was not a risk factor for CAD in this cohort[50].
DISCOVERY OF GENETIC RISK VARIANTS
The search for genetic risk variants predisposing to CAD stimulated the need for an international collaboration, which would pool resources and expertise and provide the required large sample size. This leads to the formation of an international consortium, Coronary Artery Disease Genome-Wide Replication and Meta Analysis (CARDIoGRAM), with an initial sample size of 82,000 cases and controls[51]. The initial effort was the discovery of thirteen novel genetic risk variants for CAD[52], followed by many others[53,54]. CARDIoGRAM was joined by Coronary Artery Disease (C4D) Genetics and became CardioGram Plus C4D[48]. While CARDIOoGRAMplusC4D remained the dominant group, several other groups contributed significantly to the pursuit[54-56]. These efforts led to the discovery of more than 200 genetic risk variants, each with genome-wide significance and confirmed in independent populations. The discovery of these genetic risk variants for CAD has been comprehensively covered in several reviews[57-61].
Several features of the genetic risk variants are listed in Table 1. They occur commonly and are located throughout the genome with only minimal risk associated with each variant. The increased relative risk for CAD per variant is about 10%. Over 80% of the variants are located in non-protein-coding regions, indicating the mechanism for mediating risk for CAD is indirect through altering the expression of protein-coding regions. Significantly, the mechanism whereby these risk variants mediate their risk for CAD is unknown in over 50%. This observation implies that other unknown factors are contributing to the etiology and pathogenesis of CAD, which, when discovered, could provide targets for the development of novel drugs. The features of DNA risk variants for CAD typify the risk variants predisposing to other common polygenic disorders such as diabetes or traits such as height[62].
Features of genetic risk variants for CAD
| 1. Genetic risk variants for CAD are due to common DNA variants | 3. More than 75% of genetic risk variants for CAD occur in nonprotein coding regions |
| 2. Each genetic risk variant for CAD imparts minimal risk (average increased relative risk of less than 10%) | 4. More than two-thirds of the genetic risk variants for CAD mediate risk independent of traditional risk factors |
GENETIC RISK SCORE IS INDEPENDENT OF THE RISK ASSOCIATED WITH A FAMILY HISTORY OF CAD
There is abundant evidence that a family history of CAD is associated with an increased risk of CAD[20-24]. How does family history compare with the recently developed Genetic Risk Score (GRS) based on the number of CAD risk variants inherited? This was directly addressed in a study by Tada et al. performed on a population of 23,595 from Malmo, Sweden. The GRS was calculated based on either 27 or 50 genetic risk variants[63]. The CAD risk determined by the genetic risk variants was independent of the risk associated with a family history of CAD. The study showed that self-reported family history and the objectively measured GRS are not redundant but complementary; thus, they cannot be substituted for each other. It was also observed in this study by Tada et al. that individuals with premature CAD with a high incidence of family history and a high GRS are at a greater risk of CAD than those with a family history and low GRS[63]. These results indicate that the GRS would be particularly helpful in detecting young asymptomatic individuals at risk for CAD who would benefit most from primary prevention. Recent studies confirm that the risk for CAD based on stratification by the GRS is independent of family history[64-66].
It is somewhat surprising that the family history and the GRS are not redundant, which could relate to several factors: (1) The family history is self-reported and not objective; (2) the family history is related to both environmental (smoking, diet) and hereditary factors; (3) there is usually no restriction on age for family history. Since 30% of all deaths are cardiovascular, a family history of heart disease is expected in those who live the expected average lifespan. A strong association between family history of heart disease and heart disease in the offspring is observed when parents die from premature CAD.
DEVELOPMENT AND EVALUATION OF THE POLYGENIC RISK SCORE
The genetic risk for CAD, now referred to as polygenic risk score (PRS), can be expressed as a single number based on the sum of the number of copies inherited from each genetic risk variant times the hazard ratio associated with that variant previously determined by a GWAS. The genetic risk variants, encrypted onto a microarray, can be used to genotype an individual’s DNA using a blood sample[67]. An initial attempt to predict CAD using only twelve genetic risk variants was less predictive than expected[68]. The additional discovery of more genetic risk variants significantly improved the power to risk-stratify for CAD events[65]. Retrospective genotyping for genetic risk variants predisposing to CAD of several large randomized placebo-controlled clinical trials assessing the effect of lowering plasma cholesterol with statin therapy has consistently shown that individuals with the higher polygenic risk score are at the highest risk for CAD and receive the most benefit from statin therapy. Mega et al. in 2015 used 27 genetic risk variants to retrospectively genotype previous randomized placebo controlled for clinic trials[69]. Stratification was based on the PRS and categorized into high, intermediate, and low genetic risk. The trials were performed to assess the effect of decreasing plasma cholesterol using statin therapy. The total sample size was 48,421 individuals. It consisted of two trials of primary prevention: JUPITER (Justification for the Use of Statins in Primary Prevention: an Intervention Trial Evaluating Rosuvastatin), ASCOT (Anglo-Scandinavian Cardiac Outcomes Trial); and two secondary prevention trials: CARE (the Cholesterol and Recurrent Events), PROVE-IT-TIMI 22 (The Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22). The individuals with the highest PRS were at the greatest risk, but also received the greatest benefit from statin therapy, whether as primary or secondary prevention. The genetic risk stratification was independent of TRFs. The number needed to treat to prevent one cardiac event in the group with high PRS was 25, versus 42 for the intermediate PRS, and 66 for the lowest PRS. In a similar clinical trial termed WOSCOPS[70] (West of Scottland Coronary Prevention Study), blood samples were genotyped with 57 genetic risk variants in a sample size of 10,456. The group with the highest PRS had a 44% reduction in cardiac events versus 24% in the remainder. The number needed to treat to prevent one cardiac event in the high PRS group was only 13 vs. 38 in all other groups. These results indicated that genetic risk stratification of CAD based on the PRS is superior and independent of TRFs.
Two recent clinical trials assessed the effect of PCSK9 inhibitors on plasma cholesterol: Further Cardiovascular Outcomes Research With PCSK9 Inhibitors in Subjects With Elevated Risk (FOURIER)[71], and Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab (ODESSEY)[72]. Genetic risk stratification in both trials shows the greatest benefit from decreasing plasma cholesterol was in the high genetic risk group. In FOURIER, with a sample size of 11,953 individuals receiving Evalocumab, had a 13% relative reduction in the group with conventional risk factors without high genetic risk, and a 31% reduction in the group with high genetic risk with or without conventional risk factors. In ODESSEY, with a sample size of 18,924, the group with the highest risk had the highest risk reduction of 37% vs. 13% reduction in the group with the lowest genetic risk.
To further evaluate the potential clinical application of PRS for CAD risk stratification, investigators took advantage of cohorts available in various Biobanks that are already phenotyped and genotyped for CAD. Abraham et al. risk-stratified five prospective cohorts, which included three from a large Finnish population survey (FINRISK) and two from the Framingham Heart Study for a combined sample size of 16,082[65]. In this study, the 10-year risk was also assessed using the Framingham Risk Score or ACC/AHA PCE which was added to the PRS and improved the 10-year risk prediction for cardiac events. In a study by
POLYGENIC RISK SCORE REDUCED BY DRUGS AND LIFESTYLE CHANGES
It is necessary to know that the genetic risk for CAD can be appropriately treated. It has been shown for decades that the genetic risk for CAD can be reduced by drug therapy. The target is usually the protein that mediates the risk designated by the gene. For example, statin therapy inhibits the rate-limiting enzyme necessary for the synthesis of cholesterol and thus specifically reduces the genetic risk associated with LDL-C. It is well known that about 65%-70% of the plasma concentration of cholesterol is under genetic control through the various enzymes required to synthesize cholesterol[75-77]. The previously mentioned studies that were retrospectively genotyped confirmed the beneficial effect of statin therapy in those at high genetic risk for CAD. Two prospective studies have also been performed assessing the effect of lifestyle changes on genetic risk following stratification based on the PRS. Khera et al. genotyped 55,685 participants[78]. A favorable lifestyle consisted of no obesity, a healthy diet, frequent exercise, and no current smoking. Conversely, an unfavorable lifestyle must include at least two of these unfavorable factors. Results of this study showed that the top 20% with the highest PRS had a 91% higher risk of cardiac events than those with a low PRS. A favorable lifestyle in the group with a high PRS exhibited a 46% lower risk for cardiac events than those with an unfavorable lifestyle. Tikkanen et al. collected a sample of 468,095 individuals provided by the UK biobank[79]. Risk stratification for CAD based on the PRS categorized the groups into low, intermediate, or high risk for CAD. This study showed that individuals with the highest PRS had the most benefit from physical fitness, with a 49% reduced risk for CAD.
ADVANTAGES OF THE PRS OVER CONVENTIONAL RISK FACTORS TO RISK STRATIFY FOR PRIMARY PREVENTION
One approach to significantly reduce the prevalence of CAD is early primary prevention. Current therapy, if administered early, can be expected to reduce the prevalence by 30% to 40%. The bottleneck is the lack of available markers in young asymptomatic individuals to select those at risk for CAD who would benefit most from primary prevention. The well-established conventional risk factors, except LDL-cholesterol, are age-dependent and often not present until the sixth or seventh decade[80].
One may ask why not treat everyone with increased plasma LDL-C. In the western world, this would be applied to most of this population. Plasma LDL-C, the main culprit in the pathogenesis of CAD, increases with age; the average plasma concentration of LDL-C in females in their 40s is 121 mg/dL, and in males 147 mg/dL[81] which is approximately twice the concentration recommended by the guidelines[82]. It is also known that only about 50%[1] of the population will experience a cardiac event, so 50% would not benefit from such therapy. The Cardiology Practice Guidelines introduced the Pooled Cohort Equation (PCE) to calculate the 10-year risk for CAD. They arbitrarily selected a 10-year risk of ≥ 7.5% as the minimal requirement to initiate primary prevention. To reach 7.5% risk, it usually requires at least two conventional risk factors, which are usually not present in individuals in their 40s or 50 s. This was an attempt by the authors of the Guidelines to select those at higher risk for CAD who would benefit most from prevention. In contrast, the PRS for CAD is established at conception and is age-independent; and it should be used to identify those at greater risk regardless of age since one’s DNA does not change throughout a lifetime.
ONGOING CLINICAL TRIAL TO TREAT THE INDIVIDUAL BASED ON GENETIC RISK SCORE
Evaluations of genetic risk for CAD have all been performed in large groups that are primarily pre-enrolled such as BioBanks. It remains to be determined whether the PRS would enable adequate separation of risk in an individual on whom prevention could be implemented. There has been no attempt to treat these individuals based on the results of the PRS. The study [Figure 2], referred to as the Genetic Risk Score for CAD (GRSC), was initiated in 2021 at the Dignity Health Care System in Phoenix, Arizona. Enrollment consists of all individuals (males and females) from age 30 years to 60 years with no known cardiac events. The genotyping is performed by the Baylor Human Genome Sequencing Center in Houston. The PRS is determined; and based on the results, the individuals are categorized into high, intermediate, and low genetic risk, as previously outlined by Roberts[83]. The genetic risk variants occur commonly such that most of the population will have 150 to 180 genetic risk variants. The categories of low, intermediate, and high are determined relative to the normal distribution of risk variants, as illustrated by the histogram in Figure 3. The histogram TRFs based on the PCE were also scored to provide a comprehensive risk assessment for CAD. The patient is counseled for the specific treatment depending on their total risk for CAD, and followed annually for 10 years.
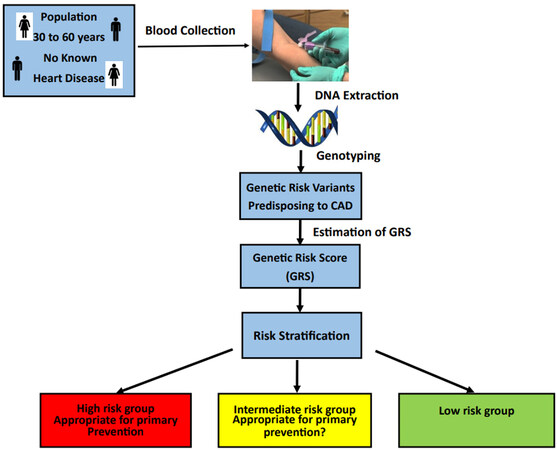
Figure 2. The population to be recruited are individuals from age 30 to 60 without known heart disease. DNA samples can be obtained from blood specimens. The DNA will be isolated and genotyped for the genetic risk variants predisposing to CAD. The genetic risk score (GRS) is calculated as a single number. Based on the GRS, the patients are stratified into three groups: high, intermediate, and low risk. Those in the high-risk group will be counseled and appropriate preventative measures will be recommended. The population will be followed annually for 10 years.
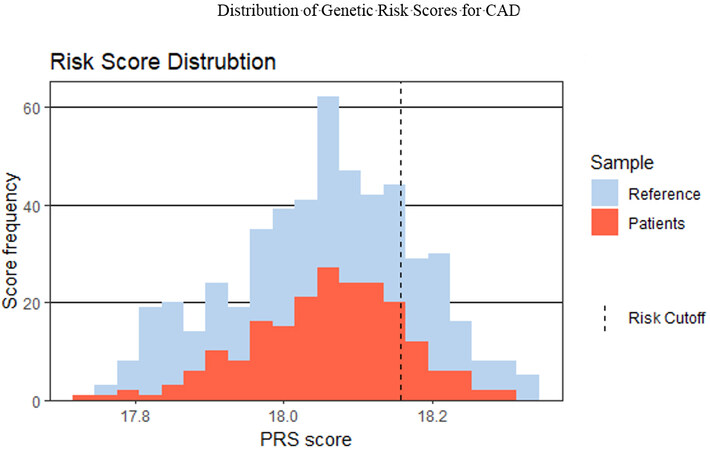
Figure 3. Binned Polygenic Risk Scores are shown on the x-axis and the frequency of occurrence in the population is shown on the y-axis. This was determined by summing the product of the number of copies of each genetic risk variant with the hazard risk ratio for CAD. Shown in blue is the distribution of the Genetic Risk Score for CAD as determined from a reference population using the same genotyping array technology and bioinformatics pipeline. Shown in orange is the distribution of the PRS scores in 205 patients genotyped as part of the ongoing clinical study, Genetic Risk Score for CAD. The risk cutoff of 18.15 was determined using the reference population and setting a threshold at the top 20%. Given the observation that these risk variants frequently occur throughout the population, it is not surprising that most of the population has many of these risk variants.
THE CAD PANDEMIC BECKONS EMBRACING GENETIC RISK STRATIFICATION
The search for more genetic risk variants continues and more will be discovered that will be expected to further improve the power of prediction. Most of the GWAS studies have been on individuals of European descent[59]. There is the concern that the application of these risk variants to other ethnic groups will be less accurate; however, a review[84] of 299 GWAS for 28 different diseases showed the disease-related genetic risk variants across the Asian and European populations were similar. This is to be expected since these variants are very common. Nevertheless, the greatest need for further GWAS is in ethnic groups and isolated populations. A recent study in East Indians by Wang et al. showed risk variants specific to this group improved genetic risk stratification for CAD based on the PRS[85]. The results of shared CAD risk variants across African and European were much less; however, the sample size was inadequate. In a study[49] comparing genetic risk variants for CAD across 2089 African Americans and 4349 Hispanics, the results were much more similar between European and Hispanics than between African American and European populations. Riveros-Mckay et al. proposed a method that combines conventional and genetic risk stratification, which was more effective than the genetic alone[86].
Several studies have confirmed the strong relationship between the intensity of the concentration[5] of plasma LDL-C and CAD. Secondly, the duration of exposure to plasma LDL-C also increases the risk such that it doubles for each additional 10 years of exposure[6]. The corollary to this observation is that early prevention is even more effective in preventing this enhancing chain of events. The proven efficacy and safety of decreasing plasma LDL-C, together with the accessibility of inexpensive therapy such as statins, should be embraced by the Current Clinical Guidelines. The cost of determining the PRS is expected to be similar to that of most conventional blood tests and centers capable of performing the analysis are widespread. Saliva, blood, or tissue specimens can be collected and shipped worldwide to an appropriate center for genotyping and analysis. Risk stratification for CAD based on the PRS would enable primary prevention at an early age. Initiating primary prevention in individuals with high genetic risk for CAD in males in their 20s or women in their 30s or 40s would be a paradigm shift in the prevention and treatment of this pandemic disease. Knowledge and technology are rapidly approaching to enable comprehensive early primary prevention of CAD, based on genetic and acquired risk, to be a worldwide endeavor.
CONCLUSION
CAD is a slowly developing disease starting in the teenage years and not manifested clinically until the sixth or seventh decade. CAD has been shown to be preventable in clinical trials primarily by decreasing traditional acquired risk factors such as hypertension. Primary prevention, which by definition is initiated early in the asymptomatic individual, is limited because it has no biomarkers to risk-stratify for CAD. This requires early prevention (30-40 years of age), but TRFs such as hypertension are age-dependent and often do not develop until the sixth or seventh decade. Genetic risk variants predisposing to CAD were first discovered in 2007 and have now increased to over 200. The genetic risk score derived from these genetic risk variants for CAD was evaluated in over one million individuals, and the prediction of cardiac risk was superior and independent of TRFs. Patients with the highest GRS had the highest risk for cardiac events and benefited most from statin therapy. Genetic risk determined at conception is not age-dependent since one’s DNA does not change in one’s lifetime. This means that the GRS can be determined at birth and used to determine primary prevention early in life. A favorable lifestyle and physical fitness in individuals with high genetic risk were shown to decrease the genetic risk for CAD by approximately 50%. The GRS, if used clinically, should significantly reduce cardiac morbidity and mortality of CAD.
DECLARATIONS
Author’s contibutionsContributed to the conception and design of the study: Roberts R
Contributed to literature search and writing: Chavira J
Contributed to Figure 3: Venner E
Availability of data and materialsNot applicable.
Financial support and sponsorshipDignity Health Foundation (455005033246), Canadian Institutes of Health Research, MOP P82810, and Canada Foundation of Innovation (11966).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
2. Lloyd-jones DM, Larson MG, Beiser A, Levy D. Lifetime risk of developing coronary heart disease. Lancet 1999;353:89-92.
3. Chan L, Boerwinkle E. Gene-environment interactions and gene therapy in atherosclerosis. Cardiol Rev 1994;2:130-7.
4. Enos WF, Holmes RH, Beyer J. Coronary disease among United States soldiers killed in action in Korea; preliminary report. J Am Med Assoc 1953;152:1090-3.
5. Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017;38:2459-72.
6. Navar-Boggan AM, Peterson ED, D’Agostino RB Sr, Neely B, Sniderman AD, Pencina MJ. Hyperlipidemia in early adulthood increases long-term risk of coronary heart disease. Circulation 2015;131:451-8.
7. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of cantos and beyond. J Am Coll Cardiol 2017;70:2278-89.
8. Cholesterol Treatment Trialists, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010;376:1670-81.
9. Collins R, Reith C, Emberson J, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016;388:2532-61.
10. Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994;344:1383-89.
11. Roberts R. PCSK9 inhibition-a new thrust in the prevention of heart disease: genetics does it again. Can J Cardiol 2013;29:899-901.
12. Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003;34:154-6.
13. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005;37:161-5.
14. Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med 2012;366:1108-18.
15. Köster A, Chao YB, Mosior M, et al. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology 2005;146:4943-50.
16. Shimamura M, Matsuda M, Yasumo H, et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol 2007;27:366-72.
17. Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med 2020;383:711-20.
18. Fujimoto K, Koishi R, Shimizugawa T, Ando Y. Angptl3-null mice show low plasma lipid concentrations by enhanced lipoprotein lipase activity. Exp Anim 2006;55:27-34.
19. Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med 2017;377:211-21.
20. Allen G, Harvald B, Shields J. Measures of twin concordance. Acta Genet Stat Med 1967;17:475-81.
21. Hopkins PN, Williams RR, Kuida H, et al. Family history as an independent risk factor for incident coronary artery disease in a high-risk cohort in Utah. Am J Cardiol 1988;62:703-7.
22. Lloyd-Jones DM, Nam BH, D’Agostino RB Sr, et al. Parental cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults: a prospective study of parents and offspring. JAMA 2004;291:2204-11.
23. Yusuf S, Hawken S, Ôunpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004;364:937-52.
24. Cooper JA, Miller GJ, Humphries SE. A comparison of the PROCAM and Framingham point-scoring systems for estimation of individual risk of coronary heart disease in the Second Northwick Park Heart Study. Atherosclerosis 2005:181,93-100.
25. Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science 1996;273:1516-7.
28. Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 2003;33:177-82.
29. Wang DG, Fan JB, Siao CJ, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 1998;280:1077-82.
30. Marian AJ, Brugada R, Roberts R. Mendelian basis of congenital and other cardiovascular diseases. In: Fuster V, Harrington RA, Narula J, Eapen ZJ, editors. Hurst’s The Heart. 14th ed. McGraw-Hill Education; 2017. Available from: https://accessmedicine.mhmedical.com/content.aspx?aid=1161732392 [Last accessed on 18 Aug 2022].
31. Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 1999;22:139-44.
32. Morgan TM, Krumholz HM, Lifton RP, Spertus JA. Nonvalidation of reported genetic risk factors for acute coronary syndrome in a large-scale replication study. JAMA 2007;297:1551-61.
33. Anand SS, Xie C, Pare G, et al. Genetic variants associated with myocardial infarction risk factors in over 8000 individuals from five ethnic groups: interheart study. Circ Cardiovas Genet 2009;2:16-25.
34. Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004;431:931-45.
35. International HapMap Consortium, Frazer KA, Ballinger DG, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007;449:851-61.
36. Stranger BE, Forrest MS, Dunning M, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007;315:848-53.
37. Roberts R. New gains in understanding coronary artery disease, interview WITH Dr Robert Roberts. Affymetrix Microarray Bulletin 2007;3:1-4.
38. Thomas DC, Haile RW, Duggan D. Recent developments in genomewide association scans: a workshop summary and review. Am J Hum Genet 2005;77:337-45.
39. Chanock SJ, Manolio T, Boehnke M, et al. Replicating genotype-phenotype associations. Nature 2007;447:655-60.
40. McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007;316:1488-91.
41. Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007;316:1491-3.
42. Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661-78.
43. O’Donnell CJ, Kavousi M, Smith AV, et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation 2011;124:2855-64.
44. Wang F, Xu CQ, He Q, et al. Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat Genet 2011;43:345-9.
45. Shen GQ, Li L, Rao S, et al. Four SNPs on chromosome 9p21 in a South Korean population implicate a genetic locus that confers high cross-race risk for development of coronary artery disease. Arterioscler Thromb Vasc Biol 2008;28:360-5.
46. Shen GQ, Rao S, Martinelli N, et al. Association between four SNPs on chromosome 9p21 and myocardial infarction is replicated in an Italian population. J Hum Genet 2007;53:144-50.
47. Hinohara K, Nakajima T, Takahashi M, et al. Replication of the association between a chromosome 9p21 polymorphism and coronary artery disease in Japanese and Korean populations. J Hum Genet 2008;53:357-9.
48. Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 2011;43:339-44.
49. Iribarren C, Lu M, Jorgenson E, et al. Weighted multi-marker genetic risk scores for incident coronary heart disease among individuals of african, latino and east-asian ancestry. Sci Rep 2018;8:6853.
50. Assimes TL, Knowles JW, Basu A, et al. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE study. Hum Mol Genet 2008;17:2320-8.
51. Preuss M, König IR, Thompson JR, et al. Design of the coronary artery disease genome-wide replication and meta-analysis (CARDIoGRAM) study: a genome-wide association meta-analysis involving more than 22,000 cases and 60,000 controls. Circ Cardiovasc Genet 2010;3:475-83.
52. Schunkert H, König IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 2011;43:333-8.
53. Deloukas P, Kanoni S, Willenborg C, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013;45:25-33.
54. Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121-30.
55. der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res 2018;122:433-43.
56. Nelson C, Goel A, Butterworth A, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 2017;49:1385-91.
58. Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet 2017;18:331-44.
59. Assimes TL, Roberts R. Genetics: implications for prevention and management of coronary artery disease. J Am Coll Cardiol 2016;68:2797-818.
60. Roberts R, Chang CC. A journey through genetic architecture and predisposition of coronary artery disease. Curr Genomics 2020;21:382-98.
61. Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res 2018;114:1241-57.
62. Nelson CP, Hamby SE, Saleheen D, et al. Genetically determined height and coronary artery disease. N Engl J Med 2015;372:1608-18.
63. Tada H, Melander O, Louie JZ, et al. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J 2016;37:561-67.
64. Schunkert H, von Scheidt M, Kessler T, Stiller B, Zeng L, Vilne B. Genetics of coronary artery disease in the light of genome-wide association studies. Clin Res Cardiol 2018;107:2-9.
65. Abraham G, Havulinna AS, Bhalala OG, et al. Genomic prediction of coronary heart disease. Eur Heart J 2016;37:3267-78.
66. Schunkert H. Family or SNPs: what counts for hereditary risk of coronary artery disease? Eur Heart J 2016;37:568-71.
67. Roberts R, Chang CC, Hadley T. Genetic risk stratification: a paradigm shift in prevention of coronary artery disease. JACC Basic Transl Sci 2021;6:287-304.
68. Davies RW, Dandona S, Stewart AF, et al. Improved prediction of cardiovascular disease based on a panel of single nucleotide polymorphisms identified through genome-wide association studies. Circ Cardiovasc Genet 2010;3:468-74.
69. Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015;385:2264-71.
70. Natarajan P, Young R, Stitziel NO, et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 2017;135:2091-101.
71. Marston NA, Kamanu FK, Nordio F, et al. Predicting benefit from evolocumab therapy in patients with atherosclerotic disease using a genetic risk score: results from the Fourier trial. Circulation 2020;141:616-23.
72. Damask A, Steg PG, Schwartz GG, et al. Patients with high genome-wide polygenic risk scores for coronary artery disease may receive greater clinical benefit from alirocumab treatment in the odyssey outcomes trial. Circulation 2020;141:624-36.
73. Inouye M, Abraham G, Nelson CP, et al. Genomic risk prediction of coronary artery disease in 480,000 adults. J Am Coll Cardiol 2018;72:1883-93.
74. Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219-24.
75. Cohen DE. Balancing cholesterol synthesis and absorption in the gastrointestinal tract. J Clin Lipidol 2008;2:S1-3.
76. Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest 2002;110:905-11.
77. Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707-13.
78. Khera AV, Emdin CA, Drake I, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med 2016;375:2349-58.
79. Tikkanen E, Gustafsson S, Ingelsson E. Associations of fitness, physical activity, strength, and genetic risk with cardiovascular disease: longitudinal analyses in the UK Biobank study. Circulation 2018;137:2583-91.
80. Roberts R, Fair J. A Less than provocative approach for the primary prevention of CAD. J Cardiovasc Transl Res 2022;15:95-102.
81. Swiger KJ, Martin SS, Blaha MJ, et al. Narrowing sex differences in lipoprotein cholesterol subclasses following mid-life: the very large database of lipids (VLDL-10B). J Am Heart Assoc 2014;3:e000851.
82. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the american college of cardiology/american heart association task force on clinical practice guidelines. Circulation 2019;139:e1082-143.
83. Roberts R. Genetic risk stratification: tipping point for global primary prevention of coronary artery disease. Circulation 2018;137:2554-6.
84. Marigorta UM, Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet 2013;9:e1003566.
85. Wang M, Menon R, Mishra S, et al. Validation of a genome-wide polygenic score for coronary artery disease in South Asians. J Am Coll Cardiol 2020;76:703-14.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Roberts R, Chavira J, Venner E. Genetic risk and its role in primary prevention of CAD. J Transl Genet Genom 2022;6:388-402. http://dx.doi.org/10.20517/jtgg.2022.07
AMA Style
Roberts R, Chavira J, Venner E. Genetic risk and its role in primary prevention of CAD. Journal of Translational Genetics and Genomics. 2022; 6(4): 388-402. http://dx.doi.org/10.20517/jtgg.2022.07
Chicago/Turabian Style
Roberts, Robert, Judith Chavira, Eric Venner. 2022. "Genetic risk and its role in primary prevention of CAD" Journal of Translational Genetics and Genomics. 6, no.4: 388-402. http://dx.doi.org/10.20517/jtgg.2022.07
ACS Style
Roberts, R.; Chavira J.; Venner E. Genetic risk and its role in primary prevention of CAD. J. Transl. Genet. Genom. 2022, 6, 388-402. http://dx.doi.org/10.20517/jtgg.2022.07
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 57 clicks
Cite This Article 57 clicks

 Like This Article 26
likes
Like This Article 26
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.