
Epigenetic effects of high-fat diet on intestinal tumorigenesis in C57BL/6J-ApcMin/+ mice

Abstract
Aim: Obesity and obesogenic diets might partly accelerate cancer development through epigenetic mechanisms. To determine these early effects, we investigated the impact of three days of a high-fat diet on epigenomic and transcriptomic changes in
Method: ChIP-Seq and RNA-Seq were performed on small intestinal epithelia of WT and
Results: Regarding epigenetic and transcriptomic changes, diet type (LFD vs. HFD) showed a significant impact, and genotype (WT vs.
Conclusion: Three days of HFD-induced epigenomic and transcriptomic changes involving metabolic and immunologic pathways that may promote tumor growth in the genetically predisposed murine intestine without affecting key cancer signaling pathways.
Keywords
INTRODUCTION
Colorectal cancer is an obesity-associated cancer where excess body fat promotes cancer development and worsens outcomes in patients with these tumors[1]. Using mouse models, we and others have shown that obesogenic diets and obesity accelerate the development of intestinal neoplasia in C57 BL6J mice with mutation or deletion of the APC gene[2,3]. Obesity has been postulated to accelerate cancer development partly through epigenetic mechanisms[4]. Murine models in which mice were fed a high-fat diet (HFD) for at least 15-20 weeks have, in fact, shown changes in histone H3 acetylation and DNA methylation associated with remodeling of chromatin regulatory regions that resemble cancer progression[5,6]. However, we have previously shown appearances of intestinal polyps in
METHODS
Mice
C57BL/6J (denoted as Apc+/+ or wild-type) and C57BL/6J-ApcMin/+ (denoted as
Experimental diets
Experimental diets consisted of a high-fat diet (HFD) and a low-fat diet (LFD), as previously described[2,9]. These diets differed in amounts of fats from hydrogenated coconut oil. The HFD contained 58.0% kcal/g of fat, 25.5% kcal/g of carbohydrate, and 16.4% kcal/g of protein (D12330; Research Diets; New Brunswick, NJ). The LFD contained 10.5% kcal/g of fat, 73.1% kcal/g of carbohydrate, and 16.4% kcal/g of protein (D12328; Research Diets; New Brunswick, NJ). Diets were otherwise matched for all micronutrients. At
Intestinal epithelia isolation
After three days on experimental diets, mice were euthanized by cervical dislocation. Small intestines were immediately removed, flushed with ice-chilled phosphate-buffered saline (PBS), and cut longitudinally. Mesenteric tissue was removed from the intestines. Each small intestine was then cut transversely into four equally sized strips. The intestinal strips were gently agitated in EDTA-based Cell Dissociation Buffer (13151014; Thermo Fisher Scientific; Waltham, MA) at 4 °C for 45 min, rinsed in cold PBS, and then agitated again in fresh dissociation buffer at 4 °C for another 45 min. Tissue strips were cut into small pieces, approximately 2 mm × 2 mm. Tissue pieces were rapidly pipetted up and down in cell dissociation buffer with a 10 mL serological pipet for 5 min or until the solution became cloudy. Intestinal pieces were removed from the solution using a 150 µm sterile paint straining mesh (2650; Mutual Dropcloth Company; Monroe, NC). The suspension was centrifuged at 200× g for 5 min, and the supernatant was aspirated, leaving a pellet of intestinal epithelia. The pellets were then suspended in 10.5 mL of cold PBS in preparation for ChIP-Seq (10 mL) and RNA-Seq (500 µL). The samples were immediately flash frozen with liquid nitrogen and stored at -80 °C for later use.
ChIP-Seq
ChIP-Seq was performed on each 10 mL intestinal epithelia sample as previously described[10]. The Covaris truChIPTM Chromatin Shearing Kit (520154; Covaris; Woburn, MA) was used to cross-link the intestinal epithelia and extract their cell nuclei according to the manufacturer’s protocol. Samples were sheared using the Covaris model S2 AFA focused ultrasonicator with the following settings: duty cycle-5%, intensity-4, cycles/burst-200, time-seven minutes). Chromatin immunoprecipitation using 9 µg of rabbit anti-H3K27ac antibody (ab4729; Abcam; Cambridge, UK) followed by recovery of DNA and then preparation of ChIP-Seq libraries were performed as previously described[11]. ChIP-Seq libraries were sequenced on an Illumina NextSeq 550 system (Illumina; San Diego, CA) at the CWRU Genomics Core.
The FASTX-Toolkit v0.013 was used to remove adapter sequences and trim read ends using a quality score cutoff of 20 (Available from: http://hannonlab.cshl.edu/fastx_toolkit/). ChIP-seq data were aligned to the mm9 genome assembly using Bowtie 2 v2.3.4.3, discarding reads with at least one mismatch and reporting the best alignment if multiple alignments were present[12]. Output SAM files were converted to binary (BAM) format, sorted, indexed, and PCR duplicates were removed using SAMtools v1.10[13]. Peaks were detected using MACS2 v2.1.2 with default settings and the broad flag set[14]. Peak lists were filtered to remove all peaks overlapping ENCODE blacklisted regions[15,16]. DeepTools v3.2.0 was used to generate RPGC-normalized bigWig tracks with 50 bp bin sizes of the final sample BAM files[17]. bigWig tracks were visualized on the Integrative Genomics Viewer to assess samples for any track irregularities or low signal-to-noise ratio[18].
Identification of variant enhancer loci
H3K27ac peaks called across samples were filtered for significance (Benjamini-Hochberg corrected
RNA-Seq
RNA was isolated from the intestinal epithelia samples using 1 mL of TRIzol (Life Technologies; Carlsbad, California; 15596-026) according to the manufacturer’s protocol by the CWRU Translational Resource Core. RNA-seq libraries were prepared and sequenced on an Illumina NextSeq 550 platform by the CWRU Genomics Core. Data were assessed for quality and trimmed for adapter sequences using Trim Galore! v0.4.2 (Babraham Bioinformatics; Cambridge, UK), a wrapper script for FastQC and Cutadapt. Reads that passed quality control were then aligned to the mouse reference genome (mm9) using the STAR aligner v2.5.3[21]. Alignment for the sequences was guided using the GENCODE annotation for mm9. Reads were analyzed for differential expression using Cufflinks v2.2.1, an RNA-Seq analysis package that reports the fragments per kilobase of exon per million fragments mapped for each gene[22]. A differential analysis report was generated using the cuffdiff command performed pairwise for each mouse group to identify differentially expressed genes (DEGs). The effect size estimate of each gene’s differential expression between the two groups was measured as log2(fold-change). Significant differential expression was identified using a cutoff of q-value < 0.05.
Functional enrichment analysis
Using the default regulatory domain definition (basal promoter 5 + 1 kb and extension up to 1 Mb), Genomic Regions Enrichment of Annotations Tool (GREAT) v4.04 was used to identify genes predicted to be associated with ChIP-Seq VELs[23]. Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) v11.5 was then used to identify biological pathways from the KEGG pathway database found to be significantly associated with these predicted genes (Benjamini-Hochberg corrected P ≤ 0.05)[24]. STRING was also used to identify pathways significantly associated with DEGs identified from RNA-Seq.
Correlation of VELs and target gene expression
DEGs predicted to be associated with VELs were identified according to the analysis of both ChIP-Seq and RNA-Seq data. The list of DEGs found from RNA-Seq was compared to the list of genes predicted by GREAT to be associated with the VELs found from ChIP-Seq. For each identical gene found in both lists, the RNA fold-change of the DEG was compared to the average H3K27ac fold-change of all of the associated VELs. VEL-gene associations in which both fold-changes were concordant (increase in RNA fold-change and increase in H3K27ac fold-change OR decrease in RNA fold-change and decrease in H3K27ac fold-change) were identified.
Hypergeometric optimization of enrichment analysis
Hypergeometric optimization of enrichment (HOMER) software was used to identify de novo motifs among the VELs (Available from: http://homer.ucsd.edu/homer/)[25]. These de novo motifs were then compared to known motifs to find the closest match to identify putative transcription factors that may be involved in interaction with the VELs.
RESULTS
High-fat diet-induced epigenomic and transcriptomic changes
H3K27ac ChIP-Seq profiles were obtained from the small intestinal epithelia samples of WT and
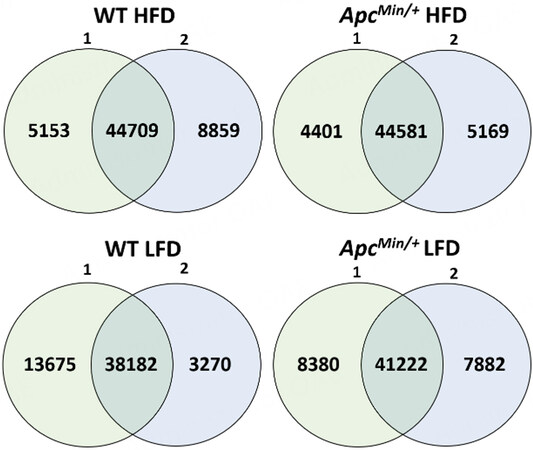
Figure 1. The number of H3K27ac peaks overlapping between the biological replicates as well as the number of peaks exclusive to each replicate for each mouse group.
A representative browser view of the H3K27ac ChIP-Seq data signal is shown in Figure 2. Gained VELs were defined as VELs with increased H3K27ac enrichment when comparing HFD profiles to LFD profiles. Lost VELs were defined as VELs with a decrease in H3K27ac enrichment when comparing HFD profiles to LFD profiles.
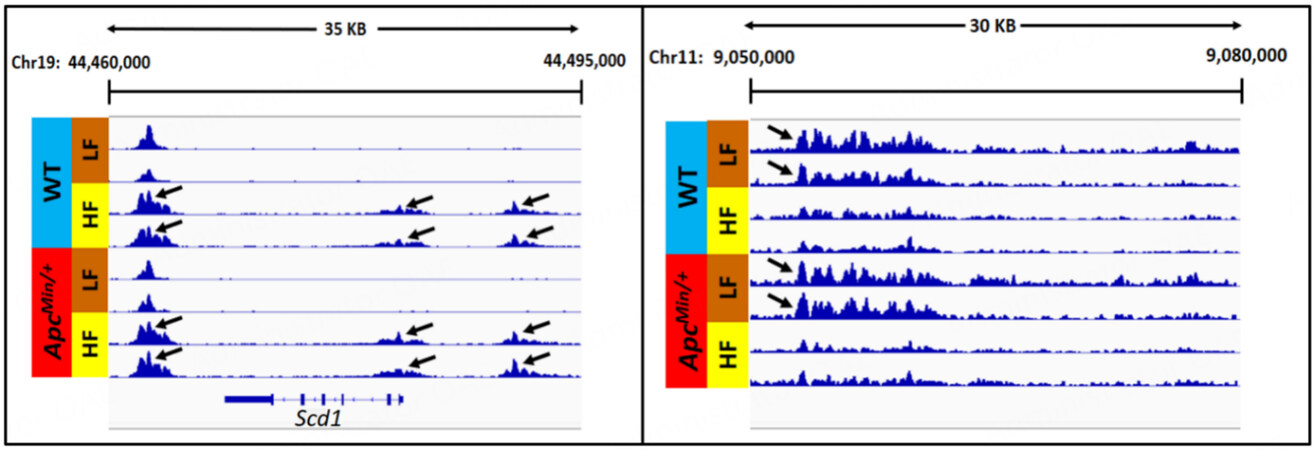
Figure 2. Normalized ChiP-Seq tracks illustrating representative examples of gained VELs (left panel) and lost VELs (right panel) induced by HF diet.
Principal component analysis showed that the H3K27ac profiles of HFD samples were highly distinct from those of LFD samples [Figure 3A]. Using BEDTools, the H3K27ac peak profiles of biological replicates were merged: 93,309 peaks were identified for WT LFD, 103,430 for WT HFD, 98,706 for
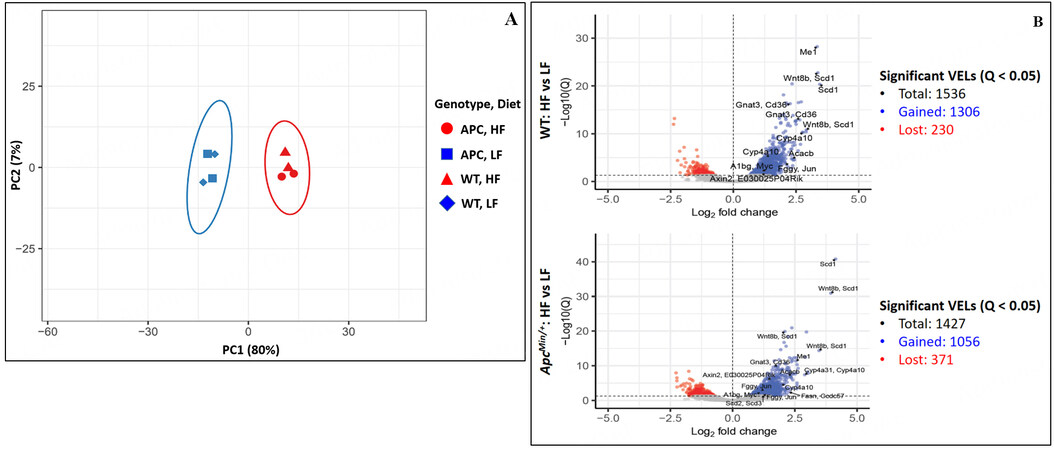
Figure 3. (A) Principal component analysis of H3K27ac profiles of the small intestinal crypt samples. (B) Total number of significant VELs identified in comparison between HFD and LFD (Bonferroni corrected P < 0.05) with certain notable VELs labeled with nearby genes. The corresponding data for these labeled VELs are shown in Supplementary Figure 6.
GREAT found multiple genes predicted to be associated with the identified VELs. In WT mice, 1660 genes were predicted to be associated with gained VELs, and 345 genes were predicted to be associated with lost VELs; In
RNA samples of adequate quality were isolated from the same epithelial samples used to obtain ChIP-Seq epigenomic profiles to generate RNA-Seq transcriptomic profiles [Supplementary Figure 1]. Similar to the epigenomic profiles, principal component analysis of the transcriptomic profiles showed a significant distinction between HFD samples and LFD samples and a less obvious distinction between similar WT samples and
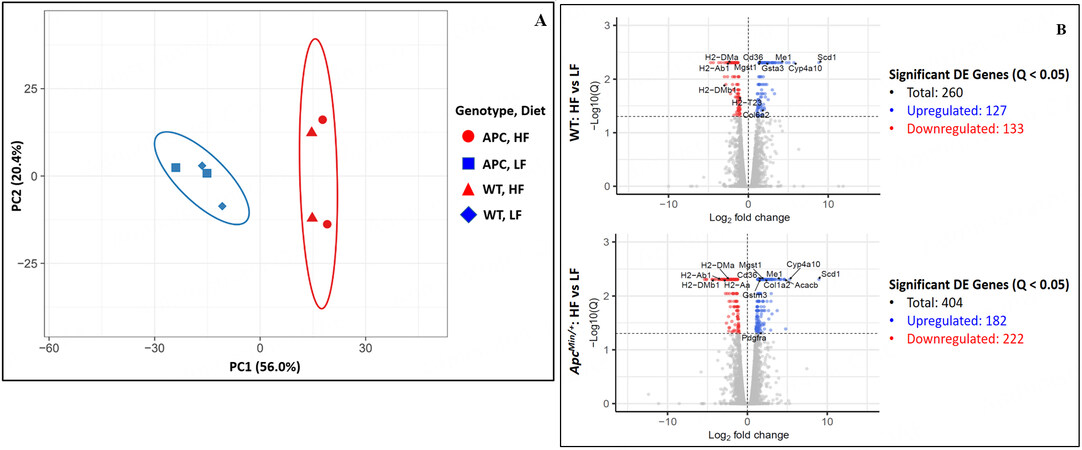
Figure 4. (A) Principal component analysis of RNA expression profiles of the small intestinal crypt samples. (B) Total number of DEGs identified in comparison between HFD to LFD (FDR Q-value < 0.05) with certain notable DEGs labeled. The corresponding data for these labeled DEGs are shown in Supplementary Figure 7.
Impact of HFD on lipid metabolism supported by epigenomic and transcriptomic changes
Multiple KEGG pathway enrichments were identified from HFD-induced gained VELs revealed by
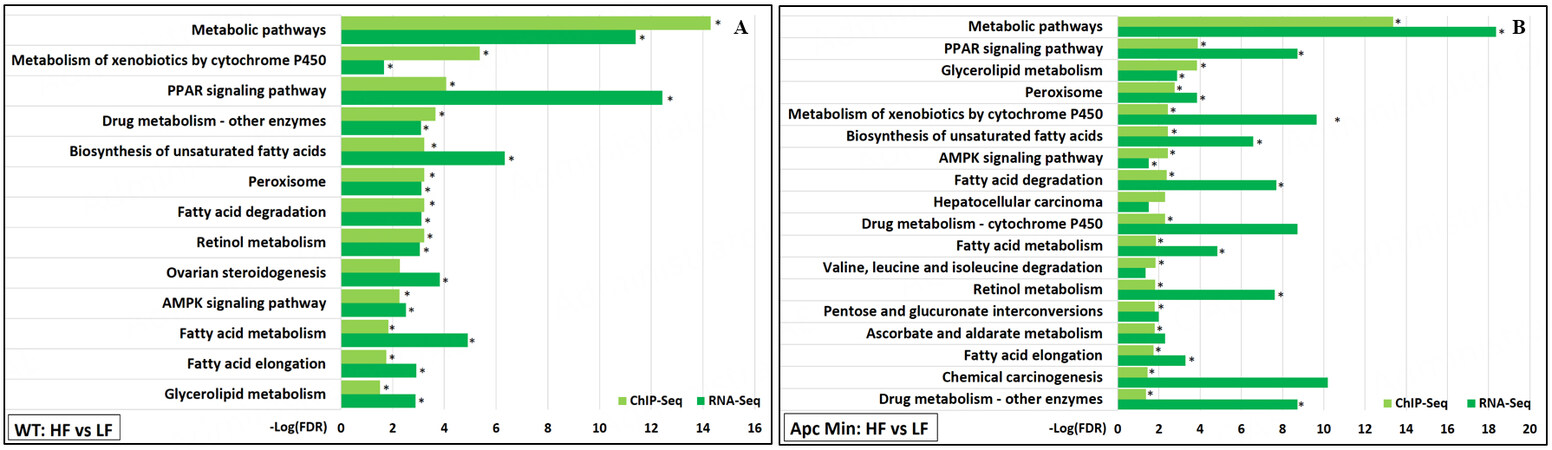
Figure 5. KEGG pathway functional enrichments significantly associated with both gained VELs (ChIP-Seq) and upregulated genes (RNA-Seq) induced by high-fat diet were identified for (A) WT mice and (B)
Although GREAT analysis of the ChIP-Seq data predicted that nearly 2000 genes were associated with gained VELs induced by HFD for each genotype, only a fraction of those genes were found to be differentially expressed according to RNA-Seq when comparing the HFD samples to the LFD samples. From analyses of the ChIP-Seq and RNA-Seq results, 76 genes upregulated in WT mice and 99 genes upregulated in
Most upregulated genes predicted to be associated with gained H3K27ac were shown to be involved in the previously mentioned lipid metabolic processes and pathways. Notably, upregulated genes included those involved in fatty acid syntheses such as fatty acid synthase (Fasn), stearoyl-CoA desaturases (Scd1, Scd2), acetyl-CoA carboxylase 2 (Acacb), and NADP-dependent malic enzyme (Me1); fatty acid oxidation such as cytochrome P450 A10 (Cyp4a10) and acetyl-Coenzyme A acyltransferase 2 (Acaa2); and fatty acid transport such as fatty acid translocase (Cd36). Overall, the ChIP-Seq and RNA-Seq analyses show evidence that the upregulation of genes involved in lipid metabolism by HFD may have a critical epigenetic mechanism.
Many epigenomic changes associated with wnt signaling
Several HFD-induced gained VELs were predicted to be associated with genes involved in the “Wnt signaling pathway”, with 41 VELs found in WT mice and 43 in
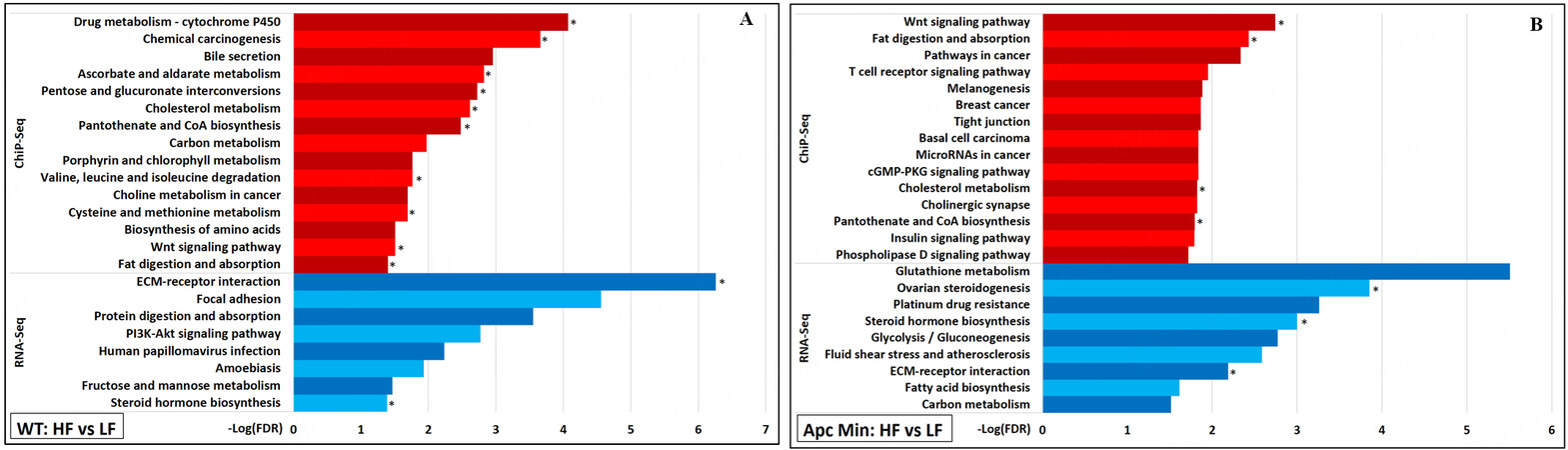
Figure 6. For (A) WT mice and for (B)
Aside from Wnt signaling, HFD was noted to upregulate the transcription of two small distinct sets of genes related to cancer. In WT mice samples, the “PI3K-Akt signaling pathway” was identified as being significantly enriched because genes encoding ligands (Col1a1, Col1a2, Col4a1, Col6a2, Col6a3, Lamb3, Tnc) and receptors (Pdgfrb, Pdgfra) that make up the “ECM-receptor interaction” and function in “focal adhesion” utilized this signaling pathway [Figure 6A][27]. Aside from Lamb3, these genes were not identified as being associated with any VELs identified from ChIP-Seq. In
HFD downregulated many genes without associated H3K27ac changes
Functional enrichment analysis of the RNA-Seq showed that several genes downregulated by HFD were associated with processes involved in the immune system [Figure 7]. There did not appear to be any lost H3K27ac correlated with the downregulation of these processes. In fact, unlike for gained VELs, no KEGG pathway enrichments were identified for lost VELs except for the “phosphatidylinositol signaling system.” The most notable immune process identified to be downregulated “antigen processing and presentation.” This process is also crucial in differentiating T cells such as Th1, Th2, and Th17[27]. Notable genes encode MHC II proteins such as H2-DMa, H2-Dmb1, and H2-Ab1; MHC I proteins such as H2-T23 and Gm11127; and the transcriptional coactivator Ciita involved in the transcription of MHC proteins.
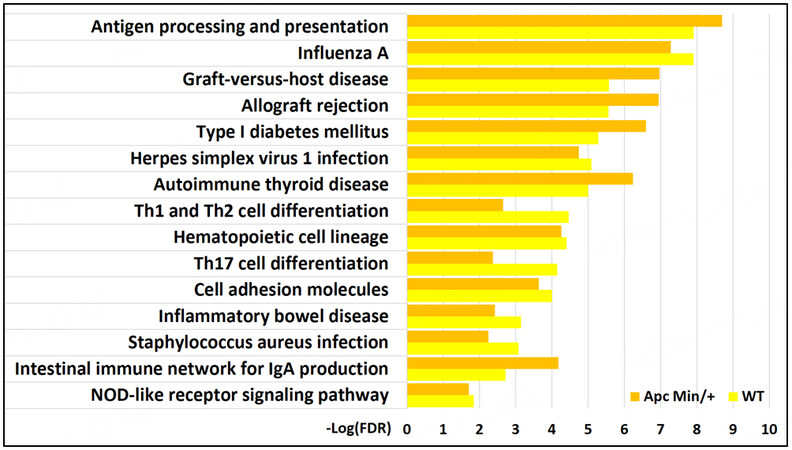
Figure 7. KEGG pathways significantly associated with genes downregulated by high-fat diet (RNA-Seq) in both WT and
Previous studies have shown that NOD-like receptors, intracellular sensors part of the innate immune response that detect compounds associated with pathogens and cell stress, are involved in MHC class II expression[30]. Genes involved in the “NOD-like receptor signaling pathway” were also found to be downregulated by an HFD in our study in WT mice (Cyba, Nfkbia, Casp4, Oas3, Ccl5, Il18) and
Predicted transcription factors associated with VELs were identified
HOMER analysis identified six likely de novo DNA motifs for WT HF vs. LF gained VELs, five for
DISCUSSION
Dysregulated lipid metabolism in cancer
Previous studies have shown that 15-20 weeks of HFD feeding resulted in multiple epigenetic changes in the intestinal epithelium[5,6]. We have previously shown that HFD promotes adenoma development in
Our studies indicate that the transcriptomic upregulation of many of these lipid metabolic genes is mediated by epigenetic changes, as shown by increased H3K27ac. Since fatty acids are essential to meet energy needs and provide the structural building blocks required for cancer cell growth, these metabolic enzymes are often upregulated in cancer[34,35]. For example, acetyl-CoA carboxylases may rewire cancer metabolism from glycolysis to lipogenesis to support energy demands for proliferation[36]. Stearoyl-CoA desaturases (SCDs) are involved in the synthesis of monounsaturated fatty acids (MUFAs) that are valuable in the formation of cancer stem cells and the promotion of their stem properties[32]. MUFAs amplify Wnt signaling via stabilization of β-catenin and LRP5/6 in rodent hepatic stellate cells and tumor-initiating cells[37]. SCD1 activity has also been shown to affect Hippo/YAP signaling by promoting the nuclear accumulation of YAP and increasing its transcriptional activity in lung adenocarcinoma cancer stem cells in a Wnt-dependent manner[38]. CD36 is commonly upregulated to facilitate increased exogenous fatty acid uptake and has been shown to play a role in tumor cell growth, metastasis, and epithelial-mesenchymal transition in multiple cancers[31]. These lipogenic genes are regulated in part by AMPK signaling[27]. AMPK signaling is essential in sensing cellular energy levels, promoting the uptake and oxidation of glucose and lipids, and inhibiting anabolic pathways that promote cell growth when energy reserves are low. Although AMPK serves as a tumor suppressor tied to energy regulation in non-cancer cells, it has been shown to act as a tumor promoter in cancer cells by protecting against various stressors, including glucose starvation and extracellular matrix detachment[39]. HOMER analysis of the gained VELs also identified a few de novo motifs similar to known nucleotide sequences that serve as binding sites for transcription factors involved in lipid metabolism (PPAR-α) and cellular proliferation (Fos, Jun-B).
Dietary suppression of immune processes
HFD downregulated several genes involved in immune processes. Among these were significant histocompatibility complex (MHC) genes involved in antigen presentation. Although MHC class II expression and function are generally considered restricted to professional antigen-presenting cells, intestinal stem cells (ISCs) located in the epithelium have also been shown to express high levels of MHC class II proteins. They can capture, process, and present antigens to CD4+ T cells[30]. Our results are substantiated by recent reports showing that HFD dampens MHC class II expression in murine intestinal epithelial cells, including intestinal stem cells[30]. Since MHC-mediated activation of CD4+ T cells is vital for tumor immunity, the loss of such MHC II expression in premalignant ISCs may enhance tumor initiation and contribute to the acceleration of tumor progression by HFD. MHC class II expression was also shown to be mediated by interferon-gamma signaling in the same study, and HFD was shown to downregulate genes involved in this pathway in our study. HOMER analysis of lost VELS also identified a few de novo motifs similar to known binding motifs for transcription factors involved in promoting immune processes (IRF, ELF3).
Role of dietary fat as a tumor promoter
Two specific cancer-promoting pathways were found to be transcriptionally upregulated after three days of HFD. Genes, predominantly ligands and receptors organizing the extracellular matrix, involved in PI3K-Akt signaling were identified to be significantly upregulated in the small intestinal epithelia of WT mice fed HFD compared to those from WT mice fed LFD. In addition, genes coding for glutathione transferases were identified to be significantly upregulated in
While dietary fat is not a tumor initiator since it is not mutagenic, its association with tumor growth suggests its role as a tumor promoter. Given the widespread H3K27ac changes induced by HFD, these results suggest a significant role for epigenetic effects in mediating the acceleration of tumor growth. However, a mutation in one or more key cancer driver genes may be required before supportive metabolic changes promote tumor growth. These observations agree with previous demonstrations that high-fat diets accelerate tumor growth in genetically predisposed tissues, i.e., with mutated oncogene or tumor-suppressed genes[40,41].
Temporal impact of HFD on the epigenome and transcriptome
When comparing this study with previous studies that fed mice HFD for 15-20 weeks, we identified differing results between the studies that suggest a temporal evolution of the epigenome and transcriptome based on the duration of HFD exposure. Li et al. showed that 15-20 weeks of HFD resulted in gained VELs associated with regulation of MAP kinase activity and JAK-STAT cascade, but these prominent cancer pathways were not enriched among gained VELs resulting from three days of HFD in our study[5]. Li et al. also found lost VELs associated with antigen presentation and processing after 15-20 weeks of HFD, whereas our study did not show these changes after three days of HFD[5]. An earlier study found transcriptional downregulation of negative regulators of several pathways involving the EGFR/RTK-RAS-ERK/MAPK cascade, TFG-β signaling, and JAK-STAT signaling; however, these negative regulators were not downregulated after three days of HFD in our study[6]. Overall, these time-dependent differences suggest a temporal progression of intestinal epigenomic and transcriptomic changes resembling cancer progression dependent on the duration of HFD exposure.
Impact of Apc min heterozygosity on epigenomic and transcriptomic changes
Our study showed that diet type had a much more significant impact on epigenomic and transcriptomic variations than the Apc Min genotype. Most of the significant processes and pathways identified from analyzing the VELs and DEGs induced by HFD were the same between each genotype. Some VELs and DEGs were identified only when comparing WT HFD and WT LFD, and some were identified only when comparing
In conclusion, Using ChIP-Seq and RNA-Seq approaches in WT C57 BL6J mice and
DECLARATIONS
AcknowledgmentsThe authors thank the CWRU Translational Resource for assisting us with RNA isolation and the CWRU Genomics Core for assisting us with preparing and sequencing our ChIP-Seq and RNA-Seq libraries.
Authors’ contributionsContributed to conception and design of study; contributed to obtaining funding for study: Berger NA, Scacheri PC, Qu DC
Contributed to preparation of materials and methods of study: Bartels CF, Hill-Baskins AE, Neu D, Qu DC, Wang R
Contributed to analysis and interpretation of study data: Chan ER, Khawaja ZQ, Lovrenert K, Neu D, Qu DC
Contributed to writing of manuscript: Berger NA, Khawaja ZQ, Qu DC
Availability of data and materialsThe data supporting the manuscript's findings are available from the corresponding author upon request.
Financial support and sponsorshipThis work was supported by the National Cancer Institute (P50CA150964, R25 CA221718, and P30CA043703 to Berger NA; R01 CA160356 and R01 CA193677 to Scacheri PC), the Clinical Translational Science Collaborative of Cleveland (UL1TR002548 to Berger NA), and the Brian Werbel Memorial Fund (Case Cancer Center Research Scholar Award to Qu DC).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateAll animal procedures were evaluated and approved by the Institutional Animal Care and Use Committee of CWRU School of Medicine, Protocol Number 2020-016.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
Supplementary MaterialsREFERENCES
1. Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body fatness and cancer - viewpoint of the IARC working group. N Engl J Med 2016;375:794-8.
2. Doerner SK, Reis ES, Leung ES, et al. High-fat diet-induced complement activation mediates intestinal inflammation and neoplasia, independent of obesity. Mol Cancer Res 2016;14:953-65.
3. Goncalves MD, Lu C, Tutnauer J, et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019;363:1345-9.
4. Berger NA, Scacheri PC. Targeting Epigenetics to prevent obesity promoted cancers. Cancer Prev Res 2018;11:125-8.
5. Li R, Grimm SA, Chrysovergis K, et al. Obesity, rather than diet, drives epigenomic alterations in colonic epithelium resembling cancer progression. Cell Metab 2014;19:702-11.
6. Li R, Grimm SA, Mav D, et al. Transcriptome and DNA methylome analysis in a mouse model of diet-induced obesity predicts increased risk of colorectal cancer. Cell Rep 2018;22:624-37.
7. Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science 2012;336:736-9.
8. Cohen AJ, Saiakhova A, Corradin O, et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat Commun 2017;8:14400.
9. Surwit R, Feinglos M, Rodin J, et al. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and mice. Metabolism 1995;44:645-51.
10. Morton AR, Dogan-Artun N, Faber ZJ, et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell 2019;179:1330-1341.e13.
11. Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, Odom DT. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods 2009;48:240-8.
12. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10:R25.
13. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078-9.
14. Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137.
15. Amemiya HM, Kundaje A, Boyle AP. The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep 2019;9:9354.
16. Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57-74.
17. Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 2014;42:W187-91.
18. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013;14:178-92.
19. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841-2.
20. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550.
21. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15-21.
22. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2013;31:46-53.
23. McLean CY, Bristor D, Hiller M, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010;28:495-501.
24. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607-13.
25. Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576-89.
26. Sethi JK, Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem J 2010;427:1-17.
27. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27-30.
28. Traverso N, Ricciarelli R, Nitti M, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev 2013;2013:972913.
29. Kennedy L, Sandhu JK, Harper ME, Cuperlovic-Culf M. Role of glutathione in cancer: from mechanisms to therapies. Biomolecules 2020;10:1429.
30. Beyaz S, Chung C, Mou H, et al. Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell 2021;28:1922-1935.e5.
31. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun 2018;38:27.
32. Li H, Feng Z, He ML. Lipid metabolism alteration contributes to and maintains the properties of cancer stem cells. Theranostics 2020;10:7053-69.
33. Yi M, Li J, Chen S, et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J Exp Clin Cancer Res 2018;37:118.
34. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer 2020;122:4-22.
35. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007;7:763-77.
36. Luo J, Hong Y, Lu Y, et al. Acetyl-CoA carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the Warburg effect by cetuximab. Cancer Lett 2017;384:39-49.
37. Lai KKY, Kweon SM, Chi F, et al. Stearoyl-CoA desaturase promotes liver fibrosis and tumor development in mice via a wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology 2017;152:1477-91.
38. Noto A, De Vitis C, Pisanu ME, et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017;36:4573-84.
39. Vara-Ciruelos D, Russell FM, Hardie DG. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol 2019;9:190099.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Qu DC, Neu D, Khawaja ZQ, Wang R, Bartels CF, Lovrenert K, Chan ER, Hill-Baskin AE, Scacheri PC, Berger NA. Epigenetic effects of high-fat diet on intestinal tumorigenesis in C57BL/6J-
AMA Style
Qu DC, Neu D, Khawaja ZQ, Wang R, Bartels CF, Lovrenert K, Chan ER, Hill-Baskin AE, Scacheri PC, Berger NA. Epigenetic effects of high-fat diet on intestinal tumorigenesis in C57BL/6J-
Chicago/Turabian Style
Qu, Dan C., Devin Neu, Zain Q. Khawaja, Ruoyu Wang, Cynthia F. Bartels, Katreya Lovrenert, Ernest R. Chan, Anne E. Hill-Baskin, Peter C. Scacheri, Nathan A. Berger. 2023. "Epigenetic effects of high-fat diet on intestinal tumorigenesis in C57BL/6J-
ACS Style
Qu, DC.; Neu D.; Khawaja ZQ.; Wang R.; Bartels CF.; Lovrenert K.; Chan ER.; Hill-Baskin AE.; Scacheri PC.; Berger NA. Epigenetic effects of high-fat diet on intestinal tumorigenesis in C57BL/6J-
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 10
likes
Like This Article 10
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.