
Genomics in practice - a review of inherited cardiac conditions


Abstract
Interest in inherited cardiovascular conditions (ICCs) is fueled by resources devoted to its diagnosis, management, and research. The rapid advancement of DNA genomic sequencing deepens our understanding of ICCs. The ICC genomic landscape empowers the development of diagnostic guidelines and the discovery of potential therapeutic targets and promises novel therapeutics, especially in precision cardiology. Therefore, it is essential for healthcare institutes and systems to develop contextual frameworks based on current guidelines to provide holistic care for patients with ICCs. The clinical frameworks and considerations described in this review provide an overview of the operations of an ICC clinic, including wet and dry lab conditions, work performed by a healthcare professional, and the variety of cases, ranging from cardiomyopathies to arrythmias to aortopathies. Insights from our experience in an ICC clinic in Singapore add to the discussion of the challenges and benefits for patients and clinicians who serve them.
Keywords
INTRODUCTION
Interest in inherited cardiovascular conditions (ICC) has risen as resources are devoted to its study[1]. Cardiovascular diseases are the most common cause of death worldwide, with numbers increasing yearly[2]. A significant proportion of these diseases are ICCs, notable for their high degree of heritable features. ICCs are broadly classified as cardiomyopathies, channelopathies, vasculopathies, heritable lipid conditions, and neuromuscular disorders involving the cardiovascular system.
The rapid advancement of DNA sequencing, from Watson and Crick’s description of the three-dimensional structure of DNA in 1953, to Sanger and Gilbert’s development of Sanger sequencing in 1977, to the most recent advanced third-generation long-read sequencing offered by Oxford Nanopore Technologies and Pacific Biosciences expands our understanding of ICCs[3]. An elucidation of the ICC genomic landscape enables the development of diagnostic guidelines, the discovery of potential therapeutic targets, and the creation of novel therapeutics. These have opened the field to precision and translational medicine in cardiology. One recent example is the US Food and Drug Administration’s approval of the first-ever drug for symptomatic obstructive hypertrophic cardiomyopathy (HCM) that works by modulating cardiac myosin[4,5].
Against this background, it is essential for healthcare institutes and systems to develop contextual frameworks making use of updated guidelines to provide holistic care for patients with ICCs[6,7]. For example, we must tackle the characterization of variants of uncertain significance (VUS), report incidental findings, navigate the myriad ethical considerations, and address the socioeconomic implications for the successful implementation of ICC genetics.
In this review, we offer insights from our experience as an ICC clinic in Singapore and introduce three case examples from our clinic.
Importance of setting up an ICC program
Current studies suggest that the prevalence of HCM, dilated cardiomyopathy (DCM), arrhythmogenic cardiomyopathy (ACM), and channelopathies are approximately 1/500, 1/250, 1/5000, and 1/2000 to 1/5000, respectively[8-12]. These are derived predominantly from studies in Western populations and extrapolated to estimate prevalence worldwide[8,11]. It is challenging to define the epidemiology due to the heterogeneity of clinical presentations and outcomes. Many ICC symptoms are shared with other common non-ICC cardiac conditions. Epidemiological estimates are likely to be conservative. One such example is HCM, where the phenotype of left ventricular hypertrophy (LVH), although sometimes accompanying unique echocardiographic features of the systolic anterior motion of the mitral valve for HCM, can be confounded by more common conditions such as hypertension or physiological LVH in athletes[13]. An additional issue is age-related penetrance or incomplete disease expression, which also hinders the diagnoses, leading to conservative estimates.
The high proportion of sudden death among the young linked to ICC[14,15] and the high prevalence of other ICCs suggests a significant burden to healthcare worldwide. To address this burden, accurate diagnosis is imperative, followed by early lifestyle and medical modifications and cascade gene testing. For example, following the diagnosis of long QT syndrome (LQTS), lifestyle changes, β-blocker therapy, left cardiac sympathetic denervation, and device therapy can be implemented early to reduce the risk of severe morbidities or sudden cardiac death (SCD)[16]. We have worked toward a systematic workflow to identify and manage ICC patients in our tertiary referral center. We follow the recommendations for a comprehensive ICC program highlighted in published genetic testing guidelines[6,7].
Overview of cases in the ICC clinic
The National University Heart Centre, Singapore (NUHCS) ICC research program was initiated in the early 2010s for patients with HCM, DCM (including arrhythmogenic right ventricular cardiomyopathy [ARVC, or now called ACM]), inherited arrhythmias and inherited aortopathies. Over time, these have been expanded to include systemic conditions with cardiac involvement, including familial hyperlipidemia and Anderson-Fabry disease. In this section, we offer a breakdown of the conditions, with relevant genetic and clinical details pertinent to the respective conditions. We only reviewed conditions that were frequently seen in our adult ICC program. For the section on inherited aortopathies, we included Marfan syndrome (MFS) because that is the most common patient we see routinely. Other aortopathies, such as Ehlers-Danlos syndrome and Loeys-Dietz syndrome, are rarely seen in our ICC program as of the writing of this manuscript; our pediatric colleagues manage them.
Inherited cardiomyopathy
Inherited cardiomyopathies form the most significant proportion of cases among our patients, with cases being classified as HCM, DCM, ARVC, and left ventricular non-compaction (LVNC).
HCM
HCM is characterized by LVH unexplained by secondary causes such as hypertension, aortic stenosis, or physiological enlargement, usually with preserved or increased ejection fraction. It is estimated to be present in 1 of 500 individuals[17]. The phenotypic presentation of HCM is heterogenous in progression and demographics, especially age, leading to conservative epidemiological statistics[18]. While most HCM cases follow a relatively benign course, HCM remains a significant cause of SCD, especially among younger patients[14,19].
Pathogenic variants in genes encoding sarcomeric proteins, including MYBPC3, MYH7, TNNT2, TNNI3, TPM1, ACTC1, MYL3, and MYL2 contribute most (up to 50%) of disease-causing variants, and these genes are commonly on HCM-specific gene panels[20,21]. Disease-causing variants contribute to varying degrees to the phenotypic presentation. For example, disease-causing TNNT2 variants are associated with poor prognosis and a high risk of SCD, while disease-causing MYBPC3 variants are associated with a delayed onset of disease and more favorable outcomes[22,23]. Risk stratification based on the mutated genes has a limited impact on patient management[18].
Substantial attention has been paid to genotype-negative HCM cases (i.e., cases lacking disease-causing variants in known HCM genes), the absence of prior family history for HCM, and the possibility of de novo HCM (cases where the variant is present in the proband but not the parents), where the disease is potentially non-familial or non-Mendelian in nature[24]. This phenomenon impacts cascade testing. In some rare instances, genotype-negative HCM cases may be explained by storage disorders (phenocopies) such as LAMP2- or PRKAG2-associated multisystem glycogen-storage disease, where the clinical presentation may not differ substantially from HCM[25].
Research is underway to establish guidelines for genotype-negative HCM cases and provide management strategies and therapeutics for the various HCM subsets. Management includes symptomatic treatment and prevention of SCD, with strategies aimed at addressing complications of the disease, such as LV obstruction or heart failure, and prophylactic measures, such as implantable cardioverter defibrillator (ICD) implantation in high-risk individuals[26,27]. It is encouraging that the first cardiac myosin inhibitor (Mavacamten) was recently approved by the United States Food and Drug Administration to treat obstructive HCM[5]. This approval opens the doors for future drugs targeting the underlying pathophysiology of HCM and (broadly) ICC in the move toward precision cardiology.
DCM
DCM is characterized by left ventricular or biventricular dilation and impaired contraction unexplained by abnormal loading conditions (e.g., hypertension and valvular heart disease) or ischemic heart disease[28]. DCM is a common cause of heart failure and a common indication for cardiac transplantation[9]. There is an estimated prevalence of 40 cases per 100,000 individuals, with differences between demographic groups such as race and age of presentation[9,29]. Notably, DCM also accounts for 60% of cardiomyopathies in childhood[30,31]. The etiology of DCM varies, with contributions commonly from genetic/familial causes and non-genetic causes. The latter includes familial DCM resulting from sarcomeric gene variants, neuromuscular disorders (e.g., Duchenne’s muscular dystrophy), inherited mitochondrial disorders, drugs (e.g., antineoplastic drugs), infections (e.g., infectious myocarditis) and others[9,32].
Familial DCM is associated with more than 50 genes, many of which encode for sarcomeric proteins and the well-known LMNA gene[33,34]. Up to 25% of familial DCM bear causal pathogenic variants in the TTN gene, whereas LMNA and MYH7 contribute 6% and 4%, respectively[32]. Genetic testing is indicated to diagnose DCM, and the diagnostic yield for genotype-positive DCM can be as high as 25%[34,35]. Interestingly, studies reported an overlap of disease-causing variants in a significant proportion of DCM patients, with 38% of 639 DCM patients having compound or combined variants with HCM and channelopathy-causing variants[36]. This finding highlights the need for precise phenotyping and additional research to understand DCM’s molecular interactions and genotype-phenotype correlations.
Genetic testing also plays some role in outcomes prediction, with some studies demonstrating a higher risk of ventricular arrhythmias and SCD in those harboring specific variants in LMNA, PLN, RBM20, and FLNC[37-42]. Another observational study reported that genotype-positive DCM patients had worse outcomes with increased rates of major adverse cardiovascular events and end-stage heart failure than genotype-negative patients[37]. Among genotype-positive DCM patients, the clinical course of DCM differs depending on the implicated gene[37]. Additional research is needed for validation and replication.
DCM management focuses on symptomatic treatment and preventing sudden death, including managing heart failure, its complications, and ICD implantation[27,28]. Currently, targeted therapeutics are being studied, with several novel therapeutic approaches, including new myosin activators[43] showing some promise. In addition to the standard therapies in the guidelines for DCM care, more are needed to advance DCM management[44].
ACM
ACM is a group of familial cell-to-cell junction cardiomyopathies underpinned by cardiac desmosome abnormalities, resulting in myocyte detachment and alteration of intracellular signal transduction. Pathological features include myocyte loss, and fibrofatty replacement of myocardium[45]. ACM predisposes patients to sustained ventricular arrhythmias, progressive ventricular dysfunction, and a high risk of SCD, especially among the young[12]. The literature estimates that the incidence of ACM is anywhere between 1/1000 and 1/5000 individuals. Guidelines suggest that the diagnosis of ACM requires a high degree of clinical suspicion with investigations such as imaging (transthoracic echocardiography or cardiac magnetic resonance imaging [cMRI]), tissue characterization (endomyocardial biopsy), repolarization and depolarization abnormalities (electrocardiography [ECG]), arrhythmia (Holter ECG monitoring), and family history[46].
Numerous pathogenic variants have been reported for ACM, with approximately half of ACM patients showing a pathogenic variant in one or more desmosome genes. Pathogenic PKP2 variants are most common, followed by pathogenic variants in DSP, DSG2, DSC2, and JUP[47,48]. Smaller subsets of ACM patients carry disease-causing variants in SCN5A, LMNA, and TTN, which are also implicated in other ICCs (i.e., DCM and Brugada syndrome [BrS]).
Genotype-negative ACM cases are similar regarding disease progression compared to genotype-positive ACM. However, different causal genes are associated with different, sometimes worse outcomes among genotype-positive ACM cases[49]. For example, pathogenic variants in PLN and DSP are more likely to be associated with heart failure[50]. In addition, a disease-causing desmosomal variant among patients with ACM often implies a worse arrhythmic course and a higher risk of SCD[38]. ACM management includes cascade testing for relatives and treatment for heart failure and arrhythmia[48].
LVNC
LVNC is a disorder of endomyocardial morphogenesis characterized by numerous and excessively prominent ventricular trabeculations and deep intertrabecular recesses; it was first described in 1984[51,52]. The diagnosis requires cMRI or echocardiography[53,54]. Its pathogenesis is not yet fully understood, explaining the lack of clarity of its genetic underpinnings. Nevertheless, because over 44% of pediatric LVNC cases are familial, it is hypothesized that gene variants disrupt the physiological compaction of the developing embryonic myocardium, resulting in LVNC[55,56].
A genomic study of LVNC implicated some genes coding for proteins of the sarcomere, Z-disk, and nuclear-envelope structures, including ACTC1, MYH7, MYBPC3, TNNT2, TPM1, TTN, LDB3, LMNA, RBM20, and DTNA[56-59]. Genotype-positive LVNC patients have poor outcomes, and closer follow-up is recommended for these patients[60,61].
There have been difficulties in establishing a consensus for the management of LVNC because of the lack of large clinical trials. Understandably, the standard of care for DCM has been extended to LVNC patients with reduced ejection fraction, with an increased focus on anticoagulation (to reduce the risk of thromboembolic stroke) and the primary prevention of SCD[62,63]. While the current management of LVNC is prophylactic and symptomatic, more is needed to improve the guidelines.
Transthyretin amyloid cardiomyopathy
Transthyretin is a ubiquitous transporter protein produced by the liver. It is a tetramer but can potentially dissociate into monomers and misfold into insoluble amyloid proteins. Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive, life-threatening disease caused by an end-organ accumulation of these misfolded transthyretin amyloid proteins, with the nerves and heart being exceptionally susceptible[64]. The disease results in a slowly progressive peripheral sensorimotor or autonomic neuropathy with progressive cardiomyopathy[65]. ATTR-CM may be associated with aging ("wild-type", wtATTR-CM, i.e., the absence of any pathogenic variants in the TTR gene) or the TTR variant, resulting in an inherent instability of the transthyretin protein (variant ATTR-CM). More than 110 TTR gene variants have been classified as pathogenic for genotype-positive ATTR-CM[66].
While not a new disease entity, ATTR-CM has received more attention in the medical community in the past decade because of the field’s rapid development. There are non-invasive diagnostics using nuclear scintigraphy that supplant the previous invasive endomyocardial biopsy[65]. More importantly, disease-modifying treatment is now available in the TTR tetramer stabilizer Tafamidis, significantly improving outcomes in patients with ATTR-CM[67].
Several registries focused on studying ATTR-CM provided many insights into this condition[68,69]. It is now widely understood that the underlying TTR variant significantly affects the clinical presentation of patients with TTR amyloidosis[66]. Variants such as NM_000371.4:c.88T>C (p.Cys30Arg) and NM_000371.4:c.349G>T (p.Ala117Ser) are associated with neurological features. Others, such as NM_000371.4:c.424G>A (p.Val142Ile), are associated with cardiac features. For the commonly encountered NM_000371.4:c.148G>A (p.Val50Met) variant, early-onset disease is associated with prominent neurological dysfunction with favorable outcomes following isolated liver transplantation, while late-onset NM_000371.4:c.148G>A (p.Val50Met) is associated with mixed neurological and cardiac dysfunction[66].
With such distinct genotype-phenotype correlations for vATTR disease presentation, accurate genotyping will enable more effective clinical management. The genotype also affects treatment decisions, especially regarding selection for liver transplantation, which is reserved mainly for early-onset NM_000371.4:c.148G>A (p.Val50Met), given the favorable outcomes of this genotype following liver transplantation[68].
Inherited channelopathies
Inherited channelopathies comprise the next most significant disease caseload in the ICC clinic. These may be spontaneously manifest or only revealed on provocation testing and systematic workup. The latter include sudden cardiac arrest (SCA) survivors with normal coronary arteries and structurally normal hearts, for whom identification of an underlying channelopathy has therapeutic and familial implications. This section covers the common inherited channelopathies: LQTS, BrS, and catecholaminergic polymorphic ventricular tachycardia (CPVT).
BrS
BrS is characterized by a coved-type ST segment elevation in the right precordial leads of an ECG and increased risk of SCD in the absence of structural abnormalities; it was first recognized in 1992 by the Brugada brothers[70]. The characteristic ECG patterns can present spontaneously or are unmasked upon provocative by sodium channel blockers such as ajmaline, procainamide, or flecainide[71]. BrS can be potentially life-threatening, with patients presenting with syncope, seizures, and nocturnal agonal breathing due to polymorphic ventricular tachycardia or ventricular fibrillation (VF). SCD may result from sustained polymorphic ventricular tachycardia or VF[71,72]. Because BrS is associated with a familial carriage, researchers have studied its genetic basis. The first genetic evidence for this condition reported in 1998 pointed to the SCN5A gene[73].
Various sodium, calcium, and potassium channel genes have been implicated in BrS pathophysiology. These include SCN4A, SCN10A, KCNH2, CACNA1C, and CACNA2D1[74]. However, only the SCN5A gene is a “definite” gene, according to an expert panel from the ClinGen consortium[75]. BrS is a monogenic Mendelian disease with an autosomal dominant inheritance pattern[74,76,77]. However, affected families frequently show incomplete penetrance, with up to 60% presenting with no family history[78]. SCN5A pathogenic variants contributed to increased severity, allowing for genetic-based risk stratification[79]. Importantly, a genome-wide association study suggested that BrS was associated with common variants, contributing to another hypothesis that BrS is a complex polygenic disease[80].
Nevertheless, genetic testing offers genetic confirmation and risk stratification[74,81]. Indeed, clinical guidelines recommend that high-risk patients receive an ICD to prevent SCD[82]. However, this decision requires careful consideration and counseling, owing to many long-term psychological sequelae aggravated by inappropriate shocks from the ICD[83]. Despite recent advances in clinical diagnoses and genetic testing, risk stratification and management remain challenging in clinical practice, not least because of the lack of clarity between the monogenic or polygenic disease causality, the frequently incomplete penetrance, and the lack of other applicable non-genetic-based stratification criteria[74,84].
LQTS
LQTS is a life-threatening inherited arrhythmia characterized by a prolonged QT interval and the onset of syncope or cardiac arrest, sometimes precipitated by emotional or physical stress[85]. LQTS can be lethal, with untreated symptomatic patients having a high mortality rate of 21% a year following syncope[86]. Mortality can be reduced to ~1% with proper and timely intervention involving a formal diagnosis, early detection, and management[16] (3, 4). Genetic testing is indicated when the clinical suspicion of LQTS is high, and the "Schwartz score" is greater than or equal to 3.5 or QTc>499 msec across multiple ECGs[87].
With advances in clinical genomics, robust phenotype-genotype correlations have been established for LQTS, making genomic analysis for this syndrome relatively more straightforward[85]. LQTS is divided into 16 different subtypes, with each subtype corresponding to a specific gene involvement[88]. By far, the first three subtypes (LQT1, LQT2, and LQT3) contribute most, corresponding to the genes KCNQ1, KCNH2, and SCN5A, respectively[89]. Among these, there is some evidence for genetic risk stratification. For example, a variant in KCNQ1, A341V, results in 80% of individuals being symptomatic, with more than 30% experiencing SCA or SCD[90]. By contrast, pathogenic variants associated with a lower frequency of SCA or death are those with LQT2 (KCNH2) and LQT3 (SCN5A) disease-causing variants, with a comparatively lower risk for life-threatening arrhythmias during exercise[85].
After the diagnosis of LQTS is established, the cornerstone for management for symptomatic patients lies with beta-blocker therapy and lifestyle modification[85]. Different or adjunctive therapeutic options can also be considered for various genetic subtypes, including sodium channel blockers for LQT3 or Andersen-Tawil syndrome (LQT7) patients[91] or calcium channel blockers for Timothy syndrome (LQT8). Other essential aspects are treating patients using left cardiac sympathetic denervation and ICD for cardiac arrest survivors[16]. While the genotype-phenotype relationship of LQTS has been extensively elucidated, understanding its complex genetic architecture might yield new management approaches.
CPVT
CPVT is a rare arrhythmogenic disorder characterized by adrenergic-induced bidirectional and polymorphic VT. The polymorphic VT tends to be reproduced during exercise, intense periods of emotions, or isoproterenol infusion and can cause syncope and SCD at a young age in the absence of structural cardiac conditions[92]. The mortality of untreated CPVT is estimated at > 31% when the patient is 30 years old, and the eight-year cardiac event rate of patients not under beta-blocker therapy is 58%[93]. CPVT is a significant cause of sudden death in the young[94]. Patients with CPVT are often missed or misdiagnosed due to the lack of obvious clinical signs or structural cardiac abnormalities, highlighting the critical role of pre-emptive genetic testing[95].
Genotype-phenotype correlations are well established for CPVT, with most carrying pathogenic RYR2 (autosomal dominant) or CASQ2 (autosomal recessive) variants. Other disease-causing variants in TRDN and TECRL have been less often implicated[92,95]. Hayashi et al. showed that cardiac and lethal event rates were similar between 50 probands and 51 affected family members[96]. This finding provides strong evidence for the importance of cascade testing in newly-diagnosed CPVT probands because affected relatives are at similarly high risk of adverse cardiac events[96].
Inherited aortopathies
MFS
MFS is an autosomal dominant multisystem connective tissue disorder associated with pathogenic variants in FBN1[97]. It is characterized by aortic root aneurysm, aortic dissection, ectopia lentis (ocular lens dislocation), and skeletal abnormalities usually involving disproportionate long bone overgrowth[98,99]. The clinical diagnosis of MFS is made using the revised Ghent Nosology, which can diagnose or exclude Marfan syndrome in 86% of cases[97,98]. With a positive diagnosis of MFS, early initiation of long-term beta-blocker therapy is essential, with angiotensin-converting enzyme receptor blockers[100]. Quinolone should be avoided because it increases the risk of aortic dissection, especially in individuals with MFS[101]. In addition to medical therapy, surgical repair is considered for significant aortic root dilation, mitral valve regurgitation, or other vascular abnormalities[100].
Up to 90% of MFS is caused by pathogenic variants in the FBN1 gene[102]. FBN1 is a large structural macromolecule found in the extracellular matrix, essential to the integrity and function of connective tissues, especially in arteries, the perichondrium, and in components of the eye. While identifying FBN1 variants is not essential for the diagnosis, it is helpful to differentiate MFS from other inheritable syndromes with similar clinical presentations[99,103]. For example, Loeys-Dietz syndrome and familial thoracic aortic aneurysms and dissections have a similar clinical presentation of skeletal abnormalities and are genetically associated with pathogenic variants in receptor genes TGFBR1 and TGFBR2 which have also been seen in MFS[104]. FBN1 variants can result in conditions other than MFS, including Weill-Marchesani syndrome, familial thoracic aortic aneurysms/dissections, acromicric dysplasia, and geleophysic dysplasia, making the clinical evaluation of the patient critical[105]. Special care must be taken when investigating individuals presenting with form fruste MFS, with atypical symptoms, or an incomplete presentation of typical MFS symptoms[106]. Genetic testing is warranted in individuals suspected to have MFS, especially when family history is unknown or ambiguous.
CLINICAL FRAMEWORK FOR PATIENTS WITH ICCs
The NUHCS ICC program was launched in 2012. This project was initiated as a research program and developed into a clinical workflow for diagnosing and managing individuals with ICC. Figure 1 summarizes the workflow for ICC evaluation.
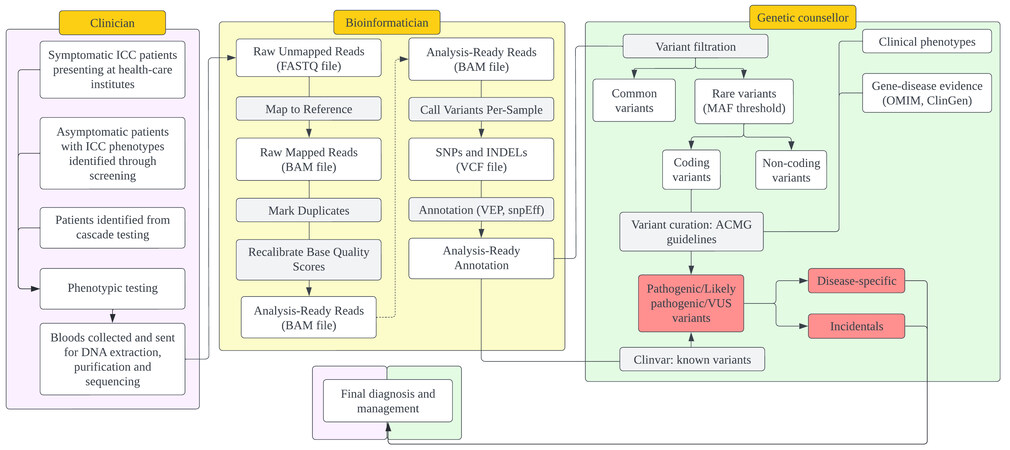
Figure 1. An overview of the process of variant-calling and gene variant curation. DNA: Deoxyribonucleic acid; SNP: single nucleotide pleomorphism; BAM: binary alignment map; INDELs: insertions and deletions; VCF: variant call format; VEP: variant effect predictor; MAF: minimum allele frequency; ACMG: American college of medical geneticists; OMIM: online mendelian inheritance in man.
Patients were identified and invited into the study if they presented symptomatically at a healthcare institute, if they were found to have phenotypes suggestive of ICC during general screening, or if they were identified as part of a cascade testing workflow. These individuals were referred to the NUHCS ICC clinic for further phenotyping and genotyping.
The phenotypic assessment was performed by clinicians using clinical examination, relevant blood tests, electrocardiography (ECG), and cardiac imaging. Cardiac imaging included echocardiography, cMRI, or computed tomography (CT) with contrast, depending on the assessed case. Following the investigations, the clinicians offered the initial explanation of the ICC to the patient and referring cardiologist/physician, with the strength of diagnosis graded according to international guidelines. Careful and accurate phenotyping is essential for solid genotype-phenotype assessments, which are also required for the genetic variant curation process.
Exome sequencing (WES) is performed for cases in the ICC clinic using next-generation sequencing, with a FASTQ file generated from the sequencing procedure. Variant calling from the FASTQ file is performed using the GATK pipeline (a bioinformatics tool that aligns the sequencing reads), presenting the overall results in a variant-called format (VCF)[107]. The VCF file contains genomic variants, categorized broadly into single nucleotide polymorphisms, insertions, and deletions. The VCF file is annotated using Variant Effect Predictor software and SnpEff[108,109]. Annotated VCF files contain details such as the variant type,
Variant curation
Variant curation is carried out by a genetic counselor trained to assess the pathogenicity of variants based on the American College of Medical Geneticists (ACMG) guidelines and, after that, counsel ICC probands and their families. As the analysis of ICC focuses on rare to ultra-rare variants with large effect sizes, the first step involves filtering variants based on allele frequency (AF) thresholds to compile for these rare coding variants. The AF thresholds for the different ICC are determined based on a model involving factors such as penetrance and population control allele frequencies[110]. AF information is compiled from international and population-specific databases of AFs with a Singapore population AF database - the SG10K_pilot database[111]. This procedure is performed so that only variants with an AF lower than maximal credible AF to suspect pathogenicity are included in the subsequent analysis because a significant pathogenicity criterion is the rarity of the variant in healthy population databases[112]. As soon as the AF threshold is implemented, the number of variants for analysis is substantially reduced.
Specific gene panels are built around published literature to associate with the patient’s presenting symptoms or suggested diagnosis. Gene panels are obtained from resources such as ClinGen and PanelApp, where experts in each field have curated disease-specific gene panels[75,113]. Genes with moderate to definite evidence of pathogenicity are included in our assessment panel, including other genes identified through our own experience [Table 1][114]. Established patterns of inheritance are also derived from international databases, consolidating available research to arrive at an expert consensus.
ICC genes are routinely used in our assessment panel consisting of genes identified through our own experience and those in Illumina Trusight panel[114]
| No. of genes | Genes |
| 176 | ACTA2, ACTC1, APOB, COL3A1, DSC2, DSG2, DSP, FBN1, GLA, KCNH2, KCNQ1, LDLR, LMNA, MYBPC3, MYH11, MYH7, MYL2, MYL3, PCSK9, PKP2, PRKAG2, RYR1, RYR2, SCN5A, SMAD3, SMAD4, TGFBR1, TGFBR2, TMEM43, TNNI3, TNNT2, TPM1, ABCC9, ABCG5, ABCG8, ACTA1, ACTN2, AKAP9, ALMS1, ANK2, ANKRD1, APOA4, APOA5, APOC2, APOE, BAG3, BRAF, CACNA1C, CACNA2D1, CACNB2, CALM1, CALM2, CALM3, CALR3, CASQ2, CAV3, CAVIN4, CBL, CBS, CETP, COL5A1, COL5A2, COX15, CREB3L3, CRELD1, CRYAB, CSRP3, CTF1, DES, DMD, DNAJC19, DOLK, DPP6, DTNA, EFEMP2, ELN, EMD, EYA4, FBN2, FHL1, FHL2, FKRP, FKTN, FXN, GAA, GATAD1, GCKR, GJA5, GPD1L, GPIHBP1, HADHA, HCN4, HFE, HRAS, HSPB8, ILK, JAG1, JPH2, JUP, KCNA5, KCND3, KCNE1, KCNE2, KCNE3, KCNJ2, KCNJ5, KCNJ8, KLF10, KRAS, LAMA2, LAMA4, LAMP2, LDB3, LDLRAP1, LMF1, LPL, LTBP2, MAP2K1, MAP2K2, MIB1, MYH6, MYLK, MYLK2, MYO6, MYOZ2, MYPN, NEXN, NKX2-5, NODAL, NOTCH1, NPPA, NRAS, PDLIM3, PLN, PRDM16, PRKAR1A, PTPN11, RAF1, RANGRF, RBM20, SALL4, SCN1B, SCN2B, SCN3B, SCN4B, SCO2, SDHA, SELENON, SGCB, SGCD, SGCG, SHOC2, SLC25A4, SLC2A10, SNTA1, SOS1, SREBF2, TAZ, TBX20, TBX3, TBX5, TCAP, TGFB2, TGFB3, TMPO, TNNC1, TRDN, TRIM63, TRPM4, TTN, TTR, TXNRD2, VCL, ZBTB17, ZHX3, ZIC3 |
The filtered variants are next curated based on other published ACMG guidelines[112]. Factors consider population data, allelic data, in silico analysis, functional data, segregation data (if available), and de novo data (if available), among others. In silico tools used to categorize and predict implications of the variant include Polyphen2[115], SIFT[116], LRT[117], MutationTaster[118], Fathmm[119], phyloP[120], CADD[121], and DANN[122]. These provide functional predictions for the variants, such as whether the amino acid substitution affects protein structure and function. Functional data include previous in vivo or in vitro studies published on specific variants and genes. To achieve high confidence, the studies must model the natural disease state as closely as possible. Family history of similar phenotypes is considered in segregation analysis, which also affects the variant classification based on the condition’s inheritance pattern. Of note, the segregation of a particular variant with a phenotype in a family is evidence for the linkage of the variant to the disorder but not any indication of the level of pathogenicity of the variant. Conversely, the absence of segregation provides evidence of the benign nature of the variant unless incomplete penetrance is considered. De novo variants are considered strong support for pathogenicity if the parents of the patients are sufficiently evaluated clinically and genetically.
All variants are cross-referenced with other curations recorded in the literature, including disease-causing variants flagged in ClinVar[123]. The variants are then analyzed in the context of the patient’s clinical history to identify (1) the disease-specific variant(s); and (2) incidental findings. To accomplish this, variants are classified according to ACMG classifications: benign, likely benign, VUS, likely pathogenic, and pathogenic.
Genome, exome, and panel sequencing
The choice of genome sequencing (WGS), exome sequencing (WES), or panel sequencing in assessing ICC considers factors such as the evidence surrounding gene-disease implications, cost-effectiveness, and depth of coverage. With recent technological advances, the cost of WGS and WES has decreased significantly without sacrificing the depth of sequencing coverage[124]. WES/WGS allows for cost-efficient diagnosis for ICC, especially for those with more than one gene implicated. WES/WGS allows for subsequent re-analysis, when additional genes can be interrogated given new research evidence or progression in the patient’s clinical condition, compared to the limited nature of gene panels[125].
Additionally, WGS/WES assesses incidental genomic findings, such as for other non-ICC conditions. Comparing WGS and WES, WGS allows for assessing the exonic and intronic genome, as opposed to WES, which covers only exonic regions. Given ever-decreasing costs, WGS may become the preferred mode of sequencing[126]. Panel sequencing may be preferred when investigating ICC with a few genes implicated, such as CPVT or LQTS, as it is targeted and results are easily interpretable. However, it lacks the benefits of WGS/WES[127].
Cascade testing
Once a disease-causing variant is identified in a proband, steps are taken for their family members to undergo similar genetic testing and phenotypic evaluation, with appropriate consent, because the patient’s immediate family members are at increased risk of harboring the same disease-causing variant[6]. From a macro perspective, cascade testing can save a healthcare system, owing to early detection and management of high-risk individuals. This sequence significantly decreases morbidity and mortality and reduces the resources required to manage these patients[128]. One signature example is familial hypercholesterolemia, where cascade testing is a proven, cost-effective means for disease early detection[129].
Cascade testing begins with the immediate family members, followed by the second-, then third-degree relatives, until all at-risk family members are identified and tested to the extent possible. The clinician and geneticist work with the proband to this end. This procedure establishes a well-annotated pedigree tree and improves the confidence of ICC diagnosis[112]. While cascade testing is critical upon discovering a disease-causing variant, there remain significant barriers and challenges to its practice. These challenges include ensuring the confidentiality of the index case (proband), the non-acceptance of such testing among family members, cultural and ethical considerations, insurance coverage, and resource considerations in these families[130]. Appropriate measures must be taken to explain the purpose and obtain informed consent before approaching family members for genetic testing[131].
Incidental secondary genetic findings
While analyzing genomic results, disease-causing variants for unrelated medical conditions may be identified incidentally. These are more likely to occur with WES/WGS sequencing. The variants may not relate to the original phenotype with which the patient presented. Relevant consent and genetic counseling must be clearly and expressly carried out before genetic testing in anticipation of these outcomes. In various cases, these incidental findings would belong under predictive diagnostic testing. Care must be applied to ensure adherence to country-specific regulations for genetic testing[132].
The ACMG lays out clear guidelines for returning incidental medically-actionable disease-causing variants in 73 genes[133]. These guidelines focus on highly penetrant genetic disorders using established management guidelines to prevent or significantly reduce mortality and morbidity. Examples of the ACMG medically-actionable incidental findings gene list include BRACA1 and BRACA2 for hereditary breast cancer, ATP7B for Wilson’s disease, and NF2 for neurofibromatosis type II[133]. Following the discovery of incidental pathogenic variants in the genes highlighted by the ACMG committee, the multidisciplinary team initiated phenotypic testing, cascade testing, and guideline-based interventions, depending on appropriate consent. Other pathogenic variants on genes not included in the ACMG list are under constant evaluation, and additional studies are required to identify the relevant incidental findings to be reported.
Patient management
After confirming a diagnosis of ICC, the long-term management of the patient and any affected family member takes the form of a multidisciplinary approach. This management begins with cardiologists and genetic counselors directing the patients’ evidence-based management, social workers assisting with long-term socioeconomic issues, and many allied health professionals. Genotype-positive and phenotype-negative family (pre-clinical) members are under increased surveillance to monitor phenotype progression[134].
Phenotype-positive, genotype-negative individuals are managed based on their clinical presentation and the latest guidelines. For specific ICCs, prognostic implications are offered. For example, better long-term outcomes are reported in genotype-negative DCM patients[37]. These are also used for decision-making, such as determining the length of time before a subsequent follow-up visit. Notably, VUS in this group of patients is subject to later reinterpretation, when the variant may become reclassified as pathogenic/likely pathogenic or benign/likely benign when new evidence appears. New research may also find that other genes are implicated in the disease condition, prompting a re-evaluation of the patient’s genotype status[6]. The latter highlights our preference for WES/WGS over panel sequencing[124].
ESSENTIAL CONSIDERATIONS FOR AN ICC CLINIC
Resource considerations in the establishment of a cardiovascular genetics program
Significant resources are required to establish a cardiovascular genetics program. In addition to the recruitment and training of a multidisciplinary team to fill the roles of cardiologists, genetic counselors, bioinformaticians, and nurse coordinators, among others, there are pertinent requirements that must be addressed for the long-term success of an ICC program. Some examples include sufficient computational power to analyze genomic information, ample database space to store genomic datasets, and a clinical and scientific framework that begins from patient recruitment and continues to cascade testing and case management. An alternative option is to outsource the process of DNA sequencing, variant-calling, annotation, and curation to an external commercial entity.
Training and education are critical for an ICC program. These include multidisciplinary meetings to ensure that team members are up-to-date with the latest guidelines and cases, journal clubs to facilitate knowledge expansion, and educational materials. Many cardiologists receive variable and often insufficient training in ICC and genomic medicine. There is also a shortage of genetic specialists, especially in less-developed jurisdictions, complicating the training in this area (8). On a positive note, the increased interest in ICCs among cardiovascular clinicians might lead to an increase in clinicians familiar with ICC diagnosis and management[135,136].
Informed consent
Bioethical considerations are essential in the practice of genomic medicine. Valid consent for genetic testing must be obtained from the patient/parent or guardian for underaged patients. They must be adequately informed regarding the details, risks, benefits, and alternatives. Consent must be voluntary and obtained by someone competent, such as a physician with relevant clinical genomics knowledge or a trained genetic counselor[131,137]. This may present challenges in less-developed countries with limited access to resources such as healthcare and education. The basis of genomics may confuse the layperson due to its highly scientific nature[138]. In addition to traditional beliefs and cost considerations, such knowledge gaps are barriers to collecting informed consent and patient’s willingness to undergo such tests[131]. The healthcare professional must be able to communicate the various implications of undergoing a genetic test, including its indications, risks, benefits, and alternatives, to ensure patient autonomy, reflecting the informed consent procedure[138]. Efforts such as providing comprehensive educational materials explaining the process help bridge the knowledge gap for patients and their families. In addition, a healthcare provider must be aware of hurdles involving the genetic testing of minors[131,138]. The provider should seek the informed consent of the minor patient’s parents (or guardian) and engage the patient in decision-making at a developmentally appropriate level, keeping with ethics guidance[131,139].
Provision of genomic services in Singapore
To ensure a responsible and comprehensive provision of clinical genetic testing, the Singapore Ministry of Health (MOH) has released a code of practice on the standards for the provision of clinical genetic and clinical laboratory genetic services. This code outlines the requirements for healthcare providers before offering genetic testing services, considering factors such as the competency levels of practitioners, types of genetic services, and clinical indications of such tests. It provides guidelines to ensure that a fully-trained provider orders genetic tests under appropriate clinical indications. These are essential because genomic testing elements such as consent taking, variant curation, and genetic counseling are complex processes and carry many implications for the patients and their families[140]. The MOH Code of Practice safeguards the interests of the healthcare provider and the patient.
Regulation of the access to genomic data
Access to genomic data and patient health data should be governed to ensure that the information is not abused. Such abuse could come in the form of unauthorized sharing of genomic information, using samples collected for purposes other than for the consented reason, or the unauthorized commercial selling of information, among others[141]. Secure databases, information security training, and regulatory bodies ensure that all data collected during these tests are managed responsibly and with sufficient accountability[142]. These factors ensure the privacy of the data and enhance public trust in the medical institutes providing genomic services. These steps go far toward assuring public receptibility to genomic testing.
Insurance (moratorium)
Genetic information can be used for many purposes, including disease prediction, disease management, or life-changing decisions. This information could also be used for insurance and employment. A key issue from the public perspective is the insurability of patients undergoing genomic testing, whether for research or medical indications. It is in the interest of insurance agencies to make use of genomic test results to assess the probability of individuals developing both rare and complex genetic diseases. This might impact the individual’s ability to buy insurance and procure insurance claims due to genetic discrimination by insurance companies. To protect their citizens, many countries have introduced legislation and agreements to minimize the risk of discrimination[143].
In Singapore, the MOH and the Life Insurance Association (LIA) have developed the "Moratorium on Genetic Testing and Insurance" to support the development of precision medicine. The LIA is a not-for-profit trade organization representing life insurance products and life reinsurance providers based in Singapore. The moratorium aims to deter individuals from undergoing clinical genetic tests for any medical indications or participating in precision medicine research due to concerns about insurability. Under this moratorium, insurance companies in Singapore are not allowed to use predictive genetic test results to assess or decide the outcome of insurance applications under various circumstances[144]. A summary of the moratorium is provided in Table 2.
Summary of the MOH - LIA moratorium protections on genetic testing and insurance. Reproduced from the MOH website: https://www.moh.gov.sg/resources-statistics/moratorium-on-genetic-testing-and-insurance, accessed 21st October 2022
| 1. Insurers are not allowed to: |
| a. Ask the applicant to take a genetic test (whether diagnostic or predictive) as part of their insurance application b. Ask the applicant to disclose and use the result of any predictive genetic test for assessing/deciding the outcome of their insurance application if the test was taken for biomedical research c. Ask applicant or medical providers to disclose, and use the result of any predictive genetic test for assessing/deciding the outcome of their insurance application if the insurance or test is any one of these: i. Health insurance, including Integrated Shield plans ii. General insurance iii. Group insurance iv. Any other insurance not covered by the Moratorium v. Direct-to-consumer genetic testing vi. Testing is done on another person (e.g., a blood relative) vii. Testing is taken after the insurance coverage had started (unless the applicant agreed to take the test before the coverage started). |
| 2. Insurers are allowed to: |
| a. Ask the applicant to disclose, and use the result of a predictive genetic test for assessing/deciding the outcome of his/her insurance application if all of the following conditions are met: i. The insurance applied for is one of the following: life, total permanent disability, long-term care, critical illness, and disability income insurance; ii. The sum assured/pay-out of insurance applied for exceeds the financial limits specified in the moratorium; iii. The applicant has taken a predictive genetic test from the list of approved predictive genetic tests specified in the moratorium for medical conditions such as Huntington’s disease and breast cancer b. Ask the applicant to disclose, and use the result of any diagnostic genetic test done for clinical care for assessing/deciding the outcome of his/her insurance application c. Use the result of any predictive genetic test (whether provided by the applicant or another person, voluntarily or accidentally, or otherwise) if the result is favorable to the applicant. |
Case studies of ICC patients from our clinic
In this section, we discuss three case examples of ICC. We present a case of cardiomyopathy, LQTS, and Marfan syndrome. These exemplify the real-life application of the workflow described in the above sections.
Case 1: A patient with DCM
The proband was a 24-year-old female at the time of presentation at the ICC clinic after a referral by her primary cardiologist given a family history of SCD. She has three brothers, one of whom passed away from SCD at age 22 shortly before the visit. Thus, she had a strong family history of DCM, with her father and deceased brother having the condition, both diagnosed through echocardiography measurements. Her family history was highly suggestive of ICC, so she was referred to the ICC clinic. [proband’s father’s echo (Supplementary Material 1)]
At the time of presentation, the proband was asymptomatic. Her transthoracic echocardiogram revealed a mildly dilated left ventricle with an ejection fraction of 52% on transthoracic echocardiography [proband’s echo (Supplementary Material 2), Figure 2A]. Due to the familial history and clinical suspicions of DCM, the proband underwent WES.

Figure 2. Echocardiographic images and a pedigree tree relevant to this proband. (A) Parasternal long axis view of a transthoracic echocardiogram. The left image belongs to the proband, and the right image belongs to the proband’s father. (B) Pedigree chart of the immediate family members. Shaded figures indicate a phenotype suggestive of ICC.
A rare variant was found on the TNNC1 gene upon genetic testing and variant curation. This gene TNNC1 is commonly implicated in DCM and HCM in an autosomal dominant inheritance pattern. The variant, in this case, was: NM_003280.3:c.452A>T (p.Asp151Val)[145]. This variant was classed as likely pathogenic by our variant curation [ACMG guidelines: PM1, PM2, PP1, PP3 (Moderate)][112]. The base position is strongly conserved, and the base change is not present in gnomAD or local databases (SG10K_pilot database) with adequate sequence coverage[111]. Pathogenic variants in the TNNC1 gene have been linked to DCM and HCM according to various databases such as OMIM, and it was labeled as "definite" according to curation by the ClinGen consortium[75]. The TNNC1 domain has 34 missense/in-frame variants (five pathogenic and 29 VUS), and the pathogenicity of the missense variant = 14.7%. In addition, clear segregation was established according to cascade testing results of the patient’s first-degree family members. In silico prediction tools, SIFT and DAMN, also offered pathogenic predictions on the variant. The variant was further confirmed through gold-standard Sanger sequencing. Eventually, cascade testing was initiated to examine other family members of the proband. The proband’s father, who bore the same variant in TNNC1, showed clinical features of DCM: dilated ventricles with a low ejection fraction of 35% [Figure 2A] and multiple hospital admissions for acute decompensated heart failure. We did not initiate testing on her deceased brother. Cascade testing for the patient’s two other brothers and mother revealed the absence of the TNNC1 variant [Figure 2B]. Clinically, they did not present with signs or symptoms of DCM or other significant cardiovascular conditions. Serial transthoracic echocardiograms of the proband’s two living brothers were normal. We took this information to represent genotype-phenotype segregation, strengthening the status of the variant as the disease-causing “pathogenic” variant.
The proband’s subsequent transthoracic echocardiogram three years after her initial presentation showed a worsening left ventricular ejection fraction of 37%. She was started on guideline-directed heart failure medications and remained asymptomatic in her follow-up visits with her primary cardiologist.
This case brings us through the entire framework of the ICC clinic, focusing on the importance of cascade testing and providing an example of how a pathogenic variant may result in heterogeneous phenotypic presentation.
Case 2: A patient with suspected LQTS
The proband was a 26-year-old female referred to the ICC clinic by her primary cardiologist given multiple episodes of arrhythmias. There was a history of two episodes of out-of-hospital VF collapse two years earlier, diagnosed as an LQTS type 2, given long QT patterns noted on the ECG [Figure 3]. Emotional outbursts seemingly triggered the two episodes. The patient was eventually fitted with a subcutaneous ICD as prophylaxis for future occurrences of VF. The patient’s DNA sample was sent for WES.
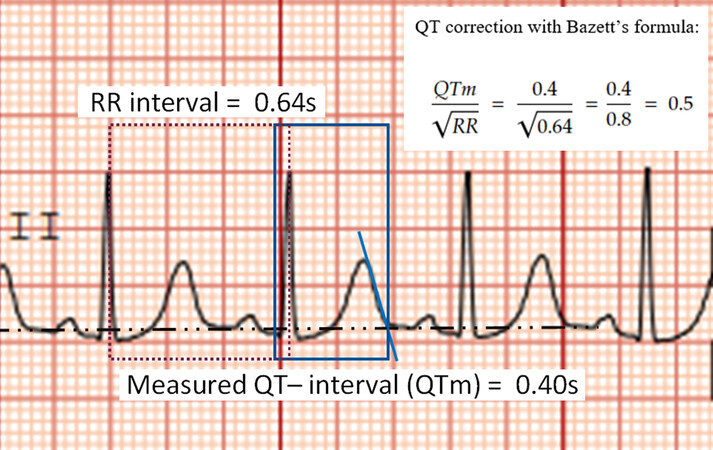
Figure 3. Lead II ECG of the proband. The calculated QTc value for this patient is 0.50 s based on this ECG (normal = 0.36-0.46 s in females).
Upon genetic testing and variant curation, no pathogenic variants were found on the genes KCNQ1, KCNH2, or SCN5A commonly implicated in LQTS. However, a likely pathogenic variant (ACMG criteria: PM1, PM2, PP2, PP3, PP5) was noted: NM_001035.3:c.14695G>A (p.Asp4899Asn). Based available from: https://www.ncbi.nlm.nih.gov/clinvar/RCV000182849.2/ 32681117 on LQTS testing guidelines, this patient was counseled on lifestyle modifications and started on anti-arrhythmic medications[16]. However, the patient presents with genotype-negative LQTS, with only one ECG showing a prolonged QT segment (other ECGs showed normal QT segments, QTc < 480 ms) and a phenotype of exertional collapse/VF; we wondered if CPVT was a differential diagnosis[146]. Pathogenic variants in RYR2 are strongly associated with CPVT, in an autosomal dominant inheritance pattern, and are less frequently associated with LQTS[146,147]. Other studies reported cases where CPVT was misdiagnosed as LQTS, with one study reporting that nearly 30% of CPVT are misdiagnosed as concealed LQTS[148-150]. The patient’s primary cardiologist was advised that a stress ECG would be required to elucidate her precise diagnosis. The proband has three older sisters and one younger sister, who are asymptomatic and have no known cardiovascular conditions. Cascade testing has been initiated for the pathogenic RYR2 variant.
This case illustrates the value of genetic testing in diagnosing inherited arrhythmias, such as BrS, CPVT, and LQTS. The test offered a means to differentiate between two conditions (LQTS and CPVT), providing the chance for a more confident diagnosis and evidence-based management for the patient.
Case 3: A patient with Marfan syndrome
This proband was a 57-year-old female patient at the time of presentation to the ICC clinic. She was referred because of her significant family history of MFS. The proband’s sister was diagnosed with MFS earlier that year, and her mother was deceased due to complications likely arising from MFS. The patient was sent for a two-dimensional echocardiogram, which revealed a dilated left ventricle with multiple regional wall motion abnormalities and moderate-to-severe aortic regurgitation. CT aortography was performed to assess her aortic root, given the high possibility that the patient has MFS. The CT aortogram showed no dissection. The aortic root z-score was calculated > 3. A diagnosis of MFS was made based on the 2010 Revised Ghent Nosology, with a positive z-score and a strong family history[98]. Genetic testing was initiated to identify the genetic basis of the patient’s condition.
Upon variant curation, a heterozygous pathogenic variant was identified on the FBN1 gene: NM_000138.5:c.4567C>T (p. Arg1523Ter). This variant has been previously annotated as pathogenic in multiple ClinVar submissions and in publications on MFS[151-154]. Variant curation also confirmed the absence of pathogenic variants in other genes, FBN2, TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, COL3A1, or COL3A2. A diagnosis of MFS was made with high confidence based on the pathogenic FBN1 variant and clinical features.
Given the diagnosis of MFS and a dilated aortic root, a modified Bentall procedure was carried out to definitively correct the aortic root dilation, and beta-blocker therapy was initiated. Cascade testing was irrelevant because there were no known extended family members, and both parents of the patient were deceased at the time of diagnosis. Five years after the bioprosthetic conduit insertion, a subaortic false aneurysm was detected on cardiac MRI during follow-up, and the patient underwent a definitive patch repair of the aneurysm [Figure 4]. Since her diagnosis and the initiation of appropriate management plans, the patient has been managing well (ten years since diagnosis).
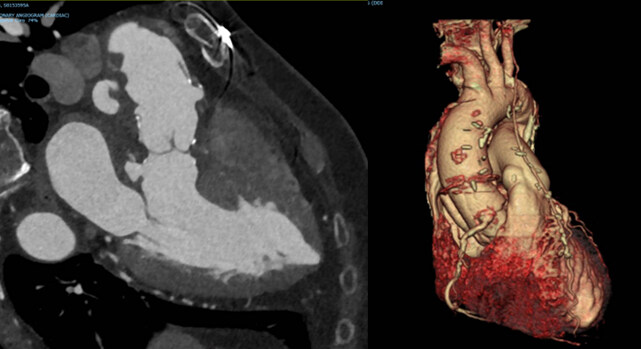
Figure 4. Radiographic images taken of the proband’s heart. Images were taken five years after the patient underwent a modified Bentall procedure. (left) MRI image of the patient’s heart post-modified Bentall procedure. (right) Three-dimensional reconstruction of the heart, using cardiac CT scans.
This case displayed the relevance of a genetic test even when clinical features and family history were clear.
CONCLUSION
The clinical framework and considerations described in this review offer clinicians and researchers an overview of what goes on in an ICC clinic and the variety of cases seen, ranging from cardiomyopathies and arrhythmias to aortopathies. While significant issues must be overcome before such a service can be implemented in healthcare centers, it is nevertheless necessary, owing to the evident burdens and needs in the community. We discussed the challenges and benefits of this service for patients and clinicians.
DECLARATIONS
Authors’ contributionsContributed to the conception and design of the study: Loong SSE, Gan LH, Klinzing DC, Tomar S, Foo RSY
Contributed to the writing of the manuscript as well as administrative, technical, and material support: Loong SSE, Gan LH, Lim YC, Ng MMQ, Wang Y, Koo SH, Chew NWS, Yeo C, Tan VH, Leong KMW, Wong RCC, Lin W, Kuntjoro I, Klinzing DC, Tomar S, Foo RSY
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe set-up of the ICC was funded by grants from the National Medical Research Council, Singapore, and NUHS, awarded to Foo RSY.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
Supplementary MaterialsREFERENCES
1. Birger M, Kaldjian AS, Roth GA, Moran AE, Dieleman JL, Bellows BK. Spending on cardiovascular disease and cardiovascular risk factors in the United States: 1996 to 2016. Circulation 2021;144:271-82.
2. Townsend N, Kazakiewicz D, Lucy Wright F, et al. Epidemiology of cardiovascular disease in Europe. Nat Rev Cardiol 2022;19:133-43.
3. Heather JM, Chain B. The sequence of sequencers: the history of sequencing DNA. Genomics 2016;107:1-8.
4. Spertus JA, Fine JT, Elliott P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): health status analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021;397:2467-75.
5. FDA. FDA approves new drug to improve heart function in adults with rare heart condition. Available from: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-new-drug-improve-heart-function-adults-rare-heart-condition [Last accessed on 20 Feb 2023].
6. Musunuru K, Hershberger RE, Day SM, et al. Genetic testing for inherited cardiovascular diseases: a scientific statement from the american heart association. Circ Genom Precis Med 2020;13:e000067.
7. Landstrom AP, Kim JJ, Gelb BD, et al. Genetic testing for heritable cardiovascular diseases in pediatric patients: a scientific statement from the american heart association. Circ Genom Precis Med 2021;14:e000086.
8. Maron BJ, McKenna WJ, Danielson GK, et al. American college of cardiology/european society of cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American college of cardiology foundation task force on clinical expert consensus documents and the European society of cardiology committee for practice guidelines. J Am Coll Cardiol 2003;42:1687-713.
10. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531-47.
11. McKenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol 2021;18:22-36.
13. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European society of cardiology (ESC). Eur Heart J 2014;35:2733-79.
14. Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980-2006. Circulation 2009;119:1085-92.
15. Podrid PJ, Myerburg RJ. Epidemiology and stratification of risk for sudden cardiac death. Clin Cardiol 2005;28:I3-11.
16. Wilde AAM, Amin AS, Postema PG. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 2022;108:332-8.
18. Mazzarotto F, Olivotto I, Boschi B, et al. Contemporary insights into the genetics of hypertrophic cardiomyopathy: toward a new era in clinical testing? J Am Heart Assoc 2020;9:e015473.
19. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 2017;121:749-70.
20. Ingles J, Goldstein J, Thaxton C, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med 2019;12:e002460.
21. Konno T, Chang S, Seidman JG, Seidman CE. Genetics of hypertrophic cardiomyopathy. Curr Opin Cardiol 2010;25:205-9.
22. Watkins H, McKenna WJ, Thierfelder L, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 1995;332:1058-64.
23. Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 1998;338:1248-57.
24. Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2018;20:69-75.
25. Arad M, Maron BJ, Gorham JM, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 2005;352:362-72.
26. Maron BJ, Desai MY, Nishimura RA, et al. Management of Hypertrophic Cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol 2022;79:390-414.
27. Maron BJ, Rowin EJ, Maron MS. Evolution of risk stratification and sudden death prevention in hypertrophic cardiomyopathy: twenty years with the implantable cardioverter-defibrillator. Heart Rhythm 2021;18:1012-23.
28. Schultheiss HP, Fairweather D, Caforio ALP, et al. Dilated cardiomyopathy. Nat Rev Dis Primers 2019;5:32.
29. Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 2006;113:1807-16.
30. Nugent AW, Daubeney PE, Chondros P, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 2003;348:1639-46.
31. Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006;296:1867-76.
32. Pinto YM, Elliott PM, Arbustini E, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 2016;37:1850-8.
33. Morales A, Hershberger RE. Genetic evaluation of dilated cardiomyopathy. Curr Cardiol Rep 2013;15:375.
34. van Spaendonck-Zwarts KY, van Rijsingen IA, van den Berg MP, et al. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years’ experience. Eur J Heart Fail 2013;15:628-36.
35. Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011;8:1308-39.
36. Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 2015;36:1123-35a.
37. Escobar-Lopez L, Ochoa JP, Mirelis JG, et al. Association of genetic variants with outcomes in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol 2021;78:1682-99.
38. Gigli M, Merlo M, Graw SL, et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol 2019;74:1480-90.
39. van den Hoogenhof MMG, Beqqali A, Amin AS, et al. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 2018;138:1330-42.
40. Kayvanpour E, Sedaghat-Hamedani F, Amr A, et al. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol 2017;106:127-39.
41. Hasselberg NE, Haland TF, Saberniak J, et al. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J 2018;39:853-60.
42. Ortiz-Genga MF, Cuenca S, Dal Ferro M, et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol 2016;68:2440-51.
43. Helms AS, Thompson AD, Day SM. Translation of new and emerging therapies for genetic cardiomyopathies. JACC Basic Transl Sci 2022;7:70-83.
44. Verdonschot JAJ, Hazebroek MR, Ware JS, Prasad SK, Heymans SRB. Role of targeted therapy in dilated cardiomyopathy: the challenging road toward a personalized approach. J Am Heart Assoc 2019;8:e012514.
45. Bosman LP, Sammani A, James CA, et al. Predicting arrhythmic risk in arrhythmogenic right ventricular cardiomyopathy: a systematic review and meta-analysis. Heart Rhythm 2018;15:1097-107.
46. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010;31:806-14.
47. van Lint FHM, Murray B, Tichnell C, et al. Arrhythmogenic right ventricular cardiomyopathy-associated desmosomal variants are rarely de novo. Circ Genom Precis Med 2019;12:e002467.
48. Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019;16:e301-72.
49. James CA, Syrris P, van Tintelen JP, Calkins H. The role of genetics in cardiovascular disease: arrhythmogenic cardiomyopathy. Eur Heart J 2020;41:1393-400.
50. Bhonsale A, Groeneweg JA, James CA, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J 2015;36:847-55.
51. Engberding R, Bender F. [Echocardiographic detection of persistent myocardial sinusoids]. Z Kardiol 1984;73:786-8.
52. Murphy RT, Thaman R, Blanes JG, et al. Natural history and familial characteristics of isolated left ventricular non-compaction. Eur Heart J 2005;26:187-92.
53. Ross SB, Jones K, Blanch B, et al. A systematic review and meta-analysis of the prevalence of left ventricular non-compaction in adults. Eur Heart J 2020;41:1428-36.
54. Bennett CE, Freudenberger R. The current approach to diagnosis and management of left ventricular noncompaction cardiomyopathy: review of the literature. Cardiol Res Pract 2016;2016:5172308.
55. Ichida F, Hamamichi Y, Miyawaki T, et al. Clinical features of isolated noncompaction of the ventricular myocardium: long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol 1999;34:233-40.
56. Sedaghat-Hamedani F, Haas J, Zhu F, et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur Heart J 2017;38:3449-60.
57. Ichida F, Tsubata S, Bowles KR, et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001;103:1256-63.
58. Probst S, Oechslin E, Schuler P, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet 2011;4:367-74.
59. Hastings R, de Villiers CP, Hooper C, et al. Combination of whole genome sequencing, linkage, and functional studies implicates a missense mutation in titin as a cause of autosomal dominant cardiomyopathy with features of left ventricular noncompaction. Circ Cardiovasc Genet 2016;9:426-35.
60. Wang C, Hata Y, Hirono K, et al. A wide and specific spectrum of genetic variants and genotype-phenotype correlations revealed by next-generation sequencing in patients with left ventricular noncompaction. J Am Heart Assoc 2017:6.
61. Li S, Zhang C, Liu N, et al. Genotype-positive status is associated with poor prognoses in patients with left ventricular noncompaction cardiomyopathy. J Am Heart Assoc 2018;7:e009910.
62. Stöllberger C, Blazek G, Dobias C, Hanafin A, Wegner C, Finsterer J. Frequency of stroke and embolism in left ventricular hypertrabeculation/noncompaction. Am J Cardiol 2011;108:1021-3.
63. Caliskan K, Szili-Torok T, Theuns DA, et al. Indications and outcome of implantable cardioverter-defibrillators for primary and secondary prophylaxis in patients with noncompaction cardiomyopathy. J Cardiovasc Electrophysiol 2011;22:898-904.
64. Sekijima Y. Hereditary transthyretin amyloidosis. In: Adam MP, Everman DB, Mirzaa GM, et al, editors. Seattle: University of Washington, 1993.
65. Maurer MS, Bokhari S, Damy T, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 2019;12:e006075.
66. Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the american heart association. Circulation 2020;142:e7-e22.
67. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007-16.
69. Damy T, Kristen AV, Suhr OB, et al. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J 2019;43:391-400.
70. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. a multicenter report. J Am Coll Cardiol 1992;20:1391-6.
71. Brugada J, Campuzano O, Arbelo E, Sarquella-Brugada G, Brugada R. Present status of brugada syndrome: JACC state-of-the-art review. J Am Coll Cardiol 2018;72:1046-59.
73. Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392:293-6.
74. Monasky MM, Micaglio E, Locati ET, Pappone C. Evaluating the use of genetics in brugada syndrome risk stratification. Front Cardiovasc Med 2021;8:652027.
75. Rehm HL, Berg JS, Brooks LD, et al. ClinGen-the clinical genome resource. N Engl J Med 2015;372:2235-42.
76. Nademanee K, Veerakul G, Chandanamattha P, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011;123:1270-9.
77. Lieve KV, Wilde AA. Inherited ion channel diseases: a brief review. Europace 2015;17 Suppl 2:ii1-6.
79. Ciconte G, Monasky MM, Santinelli V, et al. Brugada syndrome genetics is associated with phenotype severity. Eur Heart J 2021;42:1082-90.
80. Makarawate P, Glinge C, Khongphatthanayothin A, et al. Common and rare susceptibility genetic variants predisposing to Brugada syndrome in Thailand. Heart Rhythm 2020;17:2145-53.
81. Yang Y, Hu D, Sacher F, et al. Meta-analysis of risk stratification of SCN5A with brugada syndrome: is SCN5A always a marker of low risk? Front Physiol 2019;10:103.
82. Priori SG, Blomström-Lundqvist C, Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the european society of cardiology (ESC). endorsed by: association for european paediatric and congenital cardiology (AEPC). Eur Heart J 2015;36:2793-867.
83. Olde Nordkamp LR, Postema PG, Knops RE, et al. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: a systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm 2016;13:443-54.
84. Sieira J, Dendramis G, Brugada P. Pathogenesis and management of Brugada syndrome. Nat Rev Cardiol 2016;13:744-56.
85. Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol 2012;5:868-77.
87. Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013;10:1932-63.
88. Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005;33:D514-7.
89. Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 2009;6:1297-303.
90. Crotti L, Spazzolini C, Schwartz PJ, et al. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation 2007;116:2366-75.
91. Mazzanti A, Maragna R, Faragli A, et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long qt syndrome type 3. J Am Coll Cardiol 2016;67:1053-8.
92. Leenhardt A, Denjoy I, Guicheney P. Catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2012;5:1044-52.
93. Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. a 7-year follow-up of 21 patients. Circulation 1995;91:1512-9.
94. der Werf C, van Langen IM, Wilde AA. Sudden death in the young: what do we know about it and how to prevent? Circ Arrhythm Electrophysiol 2010;3:96-104.
95. Song J, Luo Y, Jiang Y, He J. Advances in the molecular genetics of catecholaminergic polymorphic ventricular tachycardia. Front Pharmacol 2021;12:718208.
96. Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 2009;119:2426-34.
98. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476-85.
101. Frankel WC, Trautner BW, Spiegelman A, Grigoryan L, LeMaire SA. Patients at risk for aortic rupture often exposed to fluoroquinolones during hospitalization. Antimicrob Agents Chemother 2019:63.
102. Coelho SG, Almeida AG. Marfan syndrome revisited: from genetics to the clinic. Rev Port Cardiol 2020;39:215-26.
103. Sakai LY, Keene DR, Renard M, De Backer J. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016;591:279-91.
104. Gao LG, Luo F, Hui RT, Zhou XL. Recent molecular biological progress in Marfan syndrome and Marfan-associated disorders. Ageing Res Rev 2010;9:363-8.
105. Cecchi A, Ogawa N, Martinez HR, et al. Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani syndrome with thoracic aortic disease and Marfan syndrome. Am J Med Genet A 2013;161A:2305-10.
106. Emanuel R, Ng RA, Marcomichelakis J, et al. Formes frustes of Marfan’s syndrome presenting with severe aortic regurgitation. clinicogenetic study of 18 families. Br Heart J 1977;39:190-7.
107. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297-303.
108. McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol 2016;17:122.
109. Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012;6:80-92.
110. Whiffin N, Minikel E, Walsh R, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med 2017;19:1151-8.
111. Wu D, Dou J, Chai X, et al. Large-scale whole-genome sequencing of three diverse asian populations in singapore. Cell 2019;179:736-749.e15.
112. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405-24.
113. Martin AR, Williams E, Foulger RE, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet 2019;51:1560-5.
114. Tomar S, Klinzing DC, Chen CK, et al. Causative variants for inherited cardiac conditions in a southeast asian population cohort. Circ Genom Precis Med 2022;15:e003536.
115. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013;7:7.20.
116. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012;40:W452-7.
117. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res 2009;19:1553-61.
118. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575-6.
119. Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 2013;34:57-65.
120. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110-21.
121. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47:D886-94.
122. Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015;31:761-3.
123. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42:D980-5.
124. Sun Y, Ruivenkamp CA, Hoffer MJ, et al. Next-generation diagnostics: gene panel, exome, or whole genome? Hum Mutat 2015;36:648-55.
125. Cirino AL, Lakdawala NK, McDonough B, et al. A comparison of whole genome sequencing to multigene panel testing in hypertrophic cardiomyopathy patients. Circ Cardiovasc Genet 2017:10.
126. Meienberg J, Bruggmann R, Oexle K, Matyas G. Clinical sequencing: is WGS the better WES? Hum Genet 2016;135:359-62.
127. Pua CJ, Bhalshankar J, Miao K, et al. Development of a Comprehensive sequencing assay for inherited cardiac condition genes. J Cardiovasc Transl Res 2016;9:3-11.
128. Sturm AC. Cardiovascular cascade genetic testing: exploring the role of direct contact and technology. Front Cardiovasc Med 2016;3:11.
129. Nherera L, Marks D, Minhas R, Thorogood M, Humphries SE. Probabilistic cost-effectiveness analysis of cascade screening for familial hypercholesterolaemia using alternative diagnostic and identification strategies. Heart 2011;97:1175-81.
130. Kam S, Bylstra Y, Forrest L, Macciocca I, Foo R. Experience of Asian males communicating cardiac genetic risk within the family. J Community Genet 2018;9:293-303.
131. McGuire AL, Beskow LM. Informed consent in genomics and genetic research. Annu Rev Genomics Hum Genet 2010;11:361-81.
132. Borry P, van Hellemondt RE, Sprumont D, et al. Legislation on direct-to-consumer genetic testing in seven European countries. Eur J Hum Genet 2012;20:715-21.
133. Miller DT, Lee K, Chung WK, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021;23:1381-90.
134. Maron BJ, Yeates L, Semsarian C. Clinical challenges of genotype positive (+)-phenotype negative (-) family members in hypertrophic cardiomyopathy. Am J Cardiol 2011;107:604-8.
135. Safarova MS, Ackerman MJ, Kullo IJ. A call for training programmes in cardiovascular genomics. Nat Rev Cardiol 2021;18:539-40.
136. Ahmad F, McNally EM, Ackerman MJ, et al. Establishment of specialized clinical cardiovascular genetics programs: recognizing the need and meeting standards: a scientific statement from the american heart association. Circ Genom Precis Med 2019;12:e000054.
137. de Vries J, Bull SJ, Doumbo O, et al. Ethical issues in human genomics research in developing countries. BMC Med Ethics 2011;12:5.
138. Girolami F, Frisso G, Benelli M, et al. Contemporary genetic testing in inherited cardiac disease: tools, ethical issues, and clinical applications. J Cardiovasc Med 2018;19:1-11.
139. Hamvas A, Madden KK, Nogee LM, et al. Informed consent for genetic research. Arch Pediatr Adolesc Med 2004;158:551-5.
140. MOH. Updates to code of practice on the standards for the provision of clinical genetic/genomic testing services and clinical laboratory genetic/genomic testing services. 2020. Available from: https://www.moh.gov.sg/docs/librariesprovider5/licensing-terms-and-conditions/moh-cir-no-234_2020_16dec20_genetic-testing.pdf [Last accessed on 20 Feb 2023].
141. Mathaiyan J, Chandrasekaran A, Davis S. Ethics of genomic research. Perspect Clin Res 2013;4:100-4.
142. Wan Z, Hazel JW, Clayton EW, Vorobeychik Y, Kantarcioglu M, Malin BA. Sociotechnical safeguards for genomic data privacy. Nat Rev Genet 2022;23:429-45.
143. Godard B, Raeburn S, Pembrey M, Bobrow M, Farndon P, Aymé S. Genetic information and testing in insurance and employment: technical, social and ethical issues. Eur J Hum Genet 2003;11 Suppl 2:S123-42.
144. MOH. Moratorium on genetic testing and insurance ministry of health, Singapore. 2021. Available from: https://www.moh.gov.sg/resources-statistics/moratorium-on-genetic-testing-and-insurance [Last accessed on 20 Feb 2023].
145. Li MX, Hwang PM. Structure and function of cardiac troponin C (TNNC1): implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015;571:153-66.
146. Letsas KP, Prappa E, Bazoukis G, et al. A novel variant of RyR2 gene in a family misdiagnosed as congenital long QT syndrome: The importance of genetic testing. J Electrocardiol 2020;60:8-11.
147. Kryshtal DO, Blackwell DJ, Egly CL, et al. RYR2 channel inhibition is the principal mechanism of flecainide action in CPVT. Circ Res 2021;128:321-31.
148. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation 1999;99:529-33.
149. Tester DJ, Kopplin LJ, Will ML, Ackerman MJ. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm 2005;2:1099-105.
150. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol 2009;54:2065-74.
151. Schrijver I, Liu W, Odom R, et al. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet 2002;71:223-37.
152. Youil R, Toner TJ, Bull E, et al. Enzymatic mutation detection (EMD) of novel mutations (R565X and R1523X) in the FBN1 gene of patients with Marfan syndrome using T4 endonuclease VII. Hum Mutat 2000;16:92-3.
153. Söylen B, Singh KK, Abuzainin A, et al. Prevalence of dural ectasia in 63 gene-mutation-positive patients with features of Marfan syndrome type 1 and Loeys-Dietz syndrome and report of 22 novel FBN1 mutations. Clin Genet 2009;75:265-70.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Loong SSE, Gan LH, Lim YC, Ng MMQ, Wang Y, Koo SH, Chew NWS, Yeo C, Tan VH, Leong KMW, Wong RCC, Lin W, Kuntjoro I, Klinzing DC, Tomar S, Foo RSY. Genomics in practice - a review of inherited cardiac conditions. J Transl Genet Genom 2023;7:27-49. http://dx.doi.org/10.20517/jtgg.2022.20
AMA Style
Loong SSE, Gan LH, Lim YC, Ng MMQ, Wang Y, Koo SH, Chew NWS, Yeo C, Tan VH, Leong KMW, Wong RCC, Lin W, Kuntjoro I, Klinzing DC, Tomar S, Foo RSY. Genomics in practice - a review of inherited cardiac conditions. Journal of Translational Genetics and Genomics. 2023; 7(1): 27-49. http://dx.doi.org/10.20517/jtgg.2022.20
Chicago/Turabian Style
Loong, Shaun Seh Ern, Louis Hanqiang Gan, Yoke Ching Lim, Miki Min Qi Ng, Yue Wang, Seok Hwee Koo, Nicholas W. S. Chew, Colin Yeo, Vern Hsen Tan, Kevin Ming Wei Leong, Raymond Ching Chiew Wong, Weiqin Lin, Ivandito Kuntjoro, David C. Klinzing, Swati Tomar, Roger S. Y. Foo. 2023. "Genomics in practice - a review of inherited cardiac conditions" Journal of Translational Genetics and Genomics. 7, no.1: 27-49. http://dx.doi.org/10.20517/jtgg.2022.20
ACS Style
Loong, SSE.; Gan LH.; Lim YC.; Ng MMQ.; Wang Y.; Koo SH.; Chew NWS.; Yeo C.; Tan VH.; Leong KMW.; Wong RCC.; Lin W.; Kuntjoro I.; Klinzing DC.; Tomar S.; Foo RSY. Genomics in practice - a review of inherited cardiac conditions. J. Transl. Genet. Genom. 2023, 7, 27-49. http://dx.doi.org/10.20517/jtgg.2022.20
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 21 clicks
Cite This Article 21 clicks

 Like This Article 15
likes
Like This Article 15
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.