
Tumor-associated antigen targets for novel immune-based strategies in prostate cancer
 0
0
Abstract
Prostate cancer remains the most common malignancy among men in the United States. Advancements in androgen receptor signaling blockade have led to landmark approvals for its use in patients with locally advanced and metastatic disease. However, additional novel therapeutic strategies for both hormone-sensitive and castration-resistant diseases remain ongoing areas of study. Thus, we turn to the growth of immuno-oncology, which has led to improved treatment outcomes for a variety of hematologic and solid tumor malignancies. Prostate cancers have shown only modest results with immune checkpoint inhibition in published trials, and innovative strategies are now looking into enhancing cytotoxic T-cell activity against cancer cells. This review provides a thorough evaluation of tumor-associated antigens that are integrated into novel chimeric antigen receptor T-cell and bispecific T-cell engager therapies. Our review will evaluate the most recent advancements in immunotherapies, while also illustrating major obstacles and underlying limiting factors.
Keywords
INTRODUCTION
Prostate cancer is the most common malignancy and the second leading cause of cancer deaths among men in the United States. In 2023, approximately 288,300 diagnoses of prostate cancer and 34,700 deaths were estimated in the United States[1,2]. Cancer incidence for prostate cancer increased by 3% annually from 2014 through 2019 after two decades of decline[2]. Due to a variety of factors including advances in therapeutics, in addition to widespread prostate-specific antigen (PSA) screening, prostate cancer mortality has declined by 53% since its peak in 1993[3,4].
Prostate cancer is an androgen-driven process[5], and thus, continuous androgen deprivation has remained the standard of care for patients with metastatic hormone-sensitive disease. Advanced understanding of testosterone suppression has led to the rise of well-known and universally used hormonal agents. While most patients initially respond to androgen suppression therapy, over time, castration resistance unavoidably occurs, leading to a need for further therapeutic options[6].
The emergence of immuno-oncology has shifted the landscape and treatment paradigm for many solid tumor malignancies[7-10]. It aims to augment the body’s innate and humoral immune response, allowing enhanced cytotoxic T-cell activity against malignant cells[10]. Despite advances in modern immunotherapy, to date, the benefits have been modest for patients with prostate cancer[11,12]. The one exception has been for mismatch-repair-deficient or microsatellite-instability-high tumors, in which pembrolizumab may be efficacious and has regulatory approval[13]. Overall, there has been limited success with immunotherapy in the treatment of prostate cancer and patients with advanced prostate cancer are in need of further developments within the immuno-oncology field. However, there is reason for optimism in immuno-oncology based on recent progress. Ongoing investigation into chimeric antigen receptor T-cell therapies (CAR-T), bispecific T-cell engagers (BiTEs), and cancer vaccines reveals their potential as novel therapies, offering patients the prospect of benefits. In this review, we outline ongoing immunotherapeutic advances in prostate cancer with a focus on novel targets for immune activation.
Targeting immune checkpoint blockade
Multiple trials have evaluated the efficacy of immune checkpoint inhibitors (ICIs) in prostate cancer
Major immune checkpoint inhibitor trials & outcomes
| Trial ID | Phase | Patients | Patients enrolled | Drug | Primary endpoint | Outcome |
| NCT00730639 (14) | Ib | mCRPC | 17 | Nivolumab | Safety tumor response Pharmacokinetics | Grade 3/4 TRAE 14% ORR 0.0% |
| NCT01375842 (15) | I | mCRPC | 35 | Atezolizumab | Safety activity | Grade 3/4 TRAE 11.4% Confirmed PSA 50 8.6% Median OS 14.7 (95%CI: 5.9-NE) months |
| NCT03016312 (16) | III | mCRPC | 759 | Atezolizumab + enzalutamide | Overall survival | Median OS 15.2 months (95%CI: 14-17) in atezolizumab + enzalutamide arm vs. OS 16.6 (95%CI: 14.7-18.4) months in enzalutamide alone |
| NCT03170960 (17) | Ib | mCRPC | 132 | Atezolizumab + cabozantinib | ORR | ORR 23%(95%CI: 17%-32%) Grade 3/4 TRAE 55% |
| NCT02484404 (18) | I/II | mCRPC | 17 | Durvalumab + olaparib | Safety PFS | Grade 3/4 TRAE 12% 12-month PFS 51.5% (95%CI: 25.7%-72.3%) |
| NCT02788773 (19) | II | mCRPC | 52 | Durvalumab +/- tremelimumab | ORR | ORR in durvalumab alone: 0% (95%CI: 0%-25%) ORR in durvalumab + tremelimumab: 16% (95%CI: 6%-32%) |
| NCT02787005 (20) | II | mCRPC | 258 | Pembrolizumab | ORR | Cohort 1: - ORR 5% (95%CI: 2%-11%) - Median OS 9.5 months Cohort 2: - ORR 3% (95%CI: < 1%-11%) - Median OS 7.9 months Cohort 3: - ORR < 1% - Median OS 14.1 months - Grade 3-5 TRAE 15% |
| NCT02787005 (21) | III | mCRPC | 126 | Pembrolizumab + enzalutamide | ORR | Cohort 4: - ORR 12.3% (95%CI: 6.1%-21.5%) - Median OS 17.6 months (95%CI: 14-22.6) - Grade 3/4 TRAE 27.2% Cohort 5: - ORR NA - Median OS 20.8 months (95%CI: 14.1-28.9) - Grade 3/4 TRAE 28.9% |
| NCT02861573 (22) | Ib/II | mCRPC | 104 | Pembrolizumab + olaparib | Safety PSA response ORR | PSA response 15% ORR 8.5% (95%CI: 2.8%-19%) Median OS 14 months (95%CI: 10.4-18.2) Grade 3-5 TRAE 48% |
| NCT01057810 (23) | III | Chemotherapy naïve mCRPC | 837 | Ipilimumab | Overall survival | Median OS 28.7 months (95%CI: 24.5-32.5) vs. placebo 29.7 months (95%CI: 26.1-34.2) Median PFS 5.6 months ipilimumab vs. 3.8 months placebo PSA response rate 23% ipilimumab vs. 8% placebo Grade 3/4 Immune-related TRAE 31% ipilimumab vs. 2% placebo |
| NCT00861614 (24) | III | mCRPC post radiation | 998 | Ipilimumab | Overall survival | Median OS 11.2 months (95%CI: 9.5-12.7) vs. 10 months (placebo) (95%CI: 8.3-11) HR 0.85 (CI 0.72-1, P = 0.053) Grade 3/4 TRAE 26% ipilimumab patients vs. 3% placebo patients |
| NCT02601014 (25) | II | mCRPC expressing AR- V7 | 30 | Ipilimumab + nivolumab | PSA response | Cohort 1: - PSA response 13% - ORR 25% - Median PFS 3 months (95%CI: 2.1-NR) - Median OS 8.2 months (95%CI: 5.5-10.4) Cohort 2: - PSA response 0% - ORR 0% - Median PFS 2.7 months (95%CI: 2.1-5.9) - Median OS 14.2 months (95%CI: 8.5-NA) - Grade 3/4 TRAE 46% in cohort 1, 53% in cohort 2 |
Targeting programmed cell death protein 1 and PD - L1
Historically, prostate cancer has been poorly responsive to targeting programmed cell death protein 1
Alternatively, durvalumab was studied in combination with olaparib. Landmark 12-month progression-free survival (PFS) was noted in 51.5%
Cytotoxic T-lymphocyte associated protein 4
Ipilimumab was studied alone and in combination with nivolumab in patients with prostate cancer. Ipilimumab monotherapy was evaluated vs. placebo in asymptomatic or minimally symptomatic chemotherapy-naïve patients with metastatic castrate-resistant prostate cancer; the evaluation did not provide any evidence of survival advantage with ipilimumab over placebo. In fact, phase III trial testing demonstrated no apparent overall survival benefit with the use of ipilimumab, the median OS of 28.7 months with ipilimumab (95%CI: 24.5-32.5 months) vs. placebo which had a median OS of 29.7 months
Ultimately, ipilimumab/nivolumab in metastatic castrate-resistant prostate cancer was deemed to have a significant risk of adverse events with minimal response[25].
TUMOR-ASSOCIATED ANTIGENS FOR T-CELL REDIRECTING THERAPIES
Despite its limited clinical success in prostate cancer, ICI therapy provided fundamental insight into the therapeutic potential of T-cell immune modulation and ushered in a new era of immunotherapies. Bispecific T-cell engager (BiTE) and chimeric antigen receptor therapies (CAR-T) are two classes of T-cell-directed immunotherapies under rapid development[26]. In contrast to ICIs, BiTE and CAR-T are MHC-independent classes of therapy that are designed to target a specific tumor-associated antigen (TAA)[27]. These therapies have immense potential to dramatically impact the treatment of prostate cancer and overcome barriers that have limited the efficacy of ICIs[28] [Figure 1].
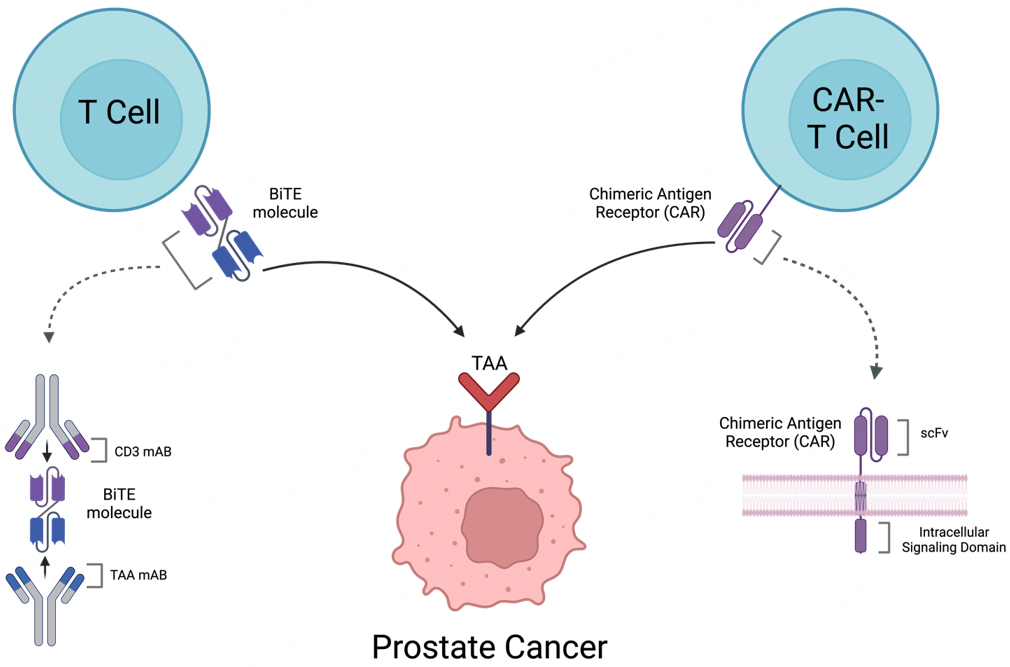
Figure 1. Schematic demonstrating the mechanism of action of BiTE & CAR-T. T-cell redirecting therapies targeting tumor-associated antigen of interest. mAB: Monoclonal antibody; scFv: single-chain variable fragment; TAA: tumor-associated antigen.
BiTEs are genetically engineered recombinant proteins with two unique single-chain variable fragments (scFvs), each with a specific monoclonal antibody. One binding region targets the CD3 region of T-cells, while the other is engineered to target a particular TAA of interest. It is important to note that this configuration allows for T-cell activation independent of the T-cell receptor-MHC pathway and circumvents a common evasion mechanism of cancer cells[27,29-32]. Simultaneous binding induces a downstream cytolytic signaling cascade, which creates the formation of a synapse between the T-cell and the target tumor cell. Subsequently, T-cells release granzymes, perforins, and cytokines, inducing cellular apoptosis of the cancer cell[33]. Furthermore, local release and diffusion of cytokines induce the upregulation of cell surface molecules on TAA-negative cells, namely the FAS death receptor, triggering further T-cell mediated lysis of tumor cells in a process termed the “bystander effect”[33,34].
CAR-T therapy is a synthetically designed receptor that is composed of three primary elements, which include an extracellular scFv with an antigen receptor specifically designed to a TAA of interest, a transmembrane domain that anchors the CAR to the T-cell membrane, and the signal transduction domain that initiates the intracellular signaling cascade within the T-cell (via CD3 and costimulatory CD28) to promote T-cell activation, proliferation, and targeted cell death. This process also occurs independently of the MHC/TCR complex[35-38]. Unlike BiTEs, CAR-T cell production is a process that is personally engineered for each individual patient, with several steps needed prior to administration. The process involves leukapheresis to isolate a patient’s T-cells, T-cell selection, transduction of these cells via a designed vector to express the CAR, expansion of the CAR-T population with growth factors, and lymphodepletion of patients prior to administration[38,39]. Once administered, the mechanism of action of CAR-T is very similar to that of a BiTE therapy, as the CAR-T molecules bind to a TAA of interest. Thereafter, cellular apoptosis is induced with the subsequent release of granzymes, perforins, and cytokines that results in sensitization of the local tumor microenvironment (TME) which has the potential to alter the immune milieu[28,35,36,40]. Despite CAR-T therapy’s revolutionary impact on hematologic malignancies, no CAR-T therapies have been approved for the treatment of solid tumor malignancies.
There is a strong biologic rationale for the employment of BiTEs and CAR-T immunotherapy in prostate cancer. MHC-independent T-cell activation avoids prostate cancer’s unique ability of MHC class I downregulation, along with its ability to prevent MHC interactions due to immunosuppressive alterations of the local TME. The immunologically “cold” TME allows prostate cancer to evade immunosurveillance and allows for continued tumor growth. However, novel immunotherapies may enhance immune cell infiltration and activation within the TME through local immunoactivation, converting the TME into a “hot” immunogenic tumor.
Clinical studies of BiTE and CAR-T therapies have demonstrated several safety concerns. Many of the established adverse effects of T-cell redirecting therapies are drawn from observations recorded from the treatment of hematologic malignancies. Notable adverse effects seen in these treatment modalities include cytokine release syndrome (CRS), immune effector-cell-associated neurotoxicity syndrome (ICANS), and on-target off-tumor (OTOT) toxicity.
CRS is a well-documented adverse event that has been observed with T-cell redirecting therapies and is characterized by an uncontrolled systemic inflammatory response as a result of high levels of pro-inflammatory cytokines released during T-cell activation, namely IL-6, IFN, and GM- CSF. CRS can present with a wide range of clinical manifestations that range from low-grade fevers to systemic organ failure[41,42]. Mild to moderate CRS is treated supportively, while severe cases are treated with infusion cessation and tocilizumab, an anti-IL-6 monoclonal antibody. Furthermore, studies have shown that CRS can be prevented with prophylactic dexamethasone administration and step-wise dose escalation[41-43].
ICANS is characterized as a neurologic toxicity associated with an increased systemic inflammatory response seen with T-cell activation. The exact pathophysiologic mechanism of ICANS remains unclear; however, it is hypothesized that activated T-cells interact and cause disruption to the blood-brain barrier and result in subsequent local CNS inflammation. Clinical manifestations are broad and can range from headache and dizziness to altered mental status and encephalopathy. Management of ICANS includes infusion cessation and steroid administration, but the efficacy of steroids remains uncertain[44-46]. Pentosan polysulfate, an antiadhesive drug, may prevent ICANS and is currently under active investigation (NCT00274742)[26,45].
OTOT toxicities occur when TAA target recognition occurs in non-neoplastic tissues and can result in cellular lysis and unintentional tissue cross-reactivity. Toxicities have been observed at higher rates in
Tumor-associated antigens as targets in immuno-oncology
Prostatic acid phosphatase
Prostatic acid phosphatase (PAP) is a glycoprotein synthesized and secreted by prostate epithelial cells and has served as one of the earliest TAAs for immunotherapy in prostate cancer, with an estimated expression of 81.8% to 91% in mCRPC[11,52,53]. Sipuleucel-T is an autologous cellular immunotherapy designed to direct the immune response towards PAP-expressing tissue. Sipuleucel-T is currently the only FDA-approved immunotherapy for the treatment of metastatic prostate cancer and serves as a category 1 NCCN treatment recommendation for patients with mCRPC who are either asymptomatic or minimally symptomatic[54]. Sipuleucel-T therapy necessitates the harvest of autologous peripheral mononuclear cells (PMC) via leukapheresis. These cells are subsequently cultured ex-vivo with a recombinant fusion protein consisting of prostate antigen and PAP linked to granulocyte- macrophage colony-stimulating factor (GM-CSF), an immune cell activator. Upon infusion into the patient, this product leads to the activation of antigen-presenting cells (APC) targeting PAP, and the PMCs, along with the activated APCs, comprise a colloquially termed “personalized cancer vaccine”[11].
The IMPACT study, which evaluated Sipuleucel-T, was a pivotal trial that played a role in the subsequent FDA approval in 2010. IMPACT was a multicenter phase III trial with 512 patients that demonstrated a 4.1-month improvement in median overall survival over placebo. AEs associated with the therapy were manageable[11,55,56]. No future confirmatory clinical trials have been performed to corroborate the findings of the IMPACT study.
Despite the observed survival benefits in metastatic castrate-resistant prostate cancer, Sipuleucel-T has failed to become a mainstay of treatment, possibly due to its complex administration and limited long-term data. However, the efficacy of Sipuleucel-T serves as proof of the principle that PAP may be an effective TAA for further immune therapy development, and autologous T-cell therapy can, to some degree, increase T-cell trafficking to tumor tissue and stimulate cytotoxic T lymphocyte activation[55].
Epithelial cell adhesion molecule
Epithelial cell adhesion molecule (EpCAM) is an epithelial cell adhesion molecule that plays an important physiological role in the formation and functionality of adhesive cellular processes, including regulation of cell-cell junctions, signaling pathways, cellular mobility, and cellular proliferation[57]. EpCAM is largely expressed in a variety of epithelial tissues including lung, colon, pancreas, bile ducts, breast tissue, and embryonic stem cells[58,59]. However, EpCAM has been found to be overexpressed in solid tumor malignancies of epithelial tissue origin, principally carcinomas[59,60]. Studies have found EpCAM expression to be 2 to 4-fold higher in mCRPC[61]. As such, EpCAM has been studied as a potential immunologic target and a focus of drug development in T-cell immunotherapies.
Among solid tumor malignancies, first-generation BiTE development was designed with EpCAM as the TAA immunologic target. Catumaxomab (Removab), a trifunctional BiTE, was the first solid tumor BiTE therapy that was approved by the European Medicines Agency in 2009 for the treatment of EpCAM-positive malignancies. Approval was based on the treatment of malignant ascites associated with epithelial cancer after a phase II/III trial of 258 patients demonstrated improvement in puncture-free survival, overall survival, and improved signs and symptoms of ascites[28,62]. Subsequent intravenous administration of catumaxomab resulted in severe AEs including cytokine release syndrome (CRS) and dose-dependent hepatotoxicity, which led to fulminant liver failure in one patient[62,63]. Ultimately, catumaxomab was voluntarily withdrawn from the market in 2017.
Solitomab (MT110 or AMG 110) is an alternative anti-EpCAM first-generation BiTE that was investigated in a phase I dose-escalation trial consisting of 65 patients with relapsed or refractory advanced-stage solid tumor malignancies known to express EpCAM, 3 (5%) of which had metastatic castrate-resistant prostate cancer. The results of the trial demonstrated grade 3 or higher AEs in over 20% of patients including severe diarrhea, abdominal pain, as well as liver enzyme and lipase elevations[63-65]. Thereafter, the study was discontinued prior to achieving dose escalation to any potentially therapeutic levels.
Currently, there are no active BiTE or CAR-T trials targeting EpCAM in prostate cancer. To some extent, the severe AEs observed are suspected to be related to OTOT cross-tissue reactivity, given the expression of EpCAM in many non-neoplastic epithelial cells. While no significant therapeutic benefits were gained from targeting EpCAM in early studies, it served as an important lesson in the development of future immunotherapy. It reinforces the importance of identifying a TAA with expression restricted to target tissue.
Prostate-specific membrane antigen
Prostate-specific membrane antigen (PSMA), or folate hydrolase 1, is a type II transmembrane protein that is composed of 750 amino acids, of which 707 are located extracellularly[66,67]. The protein is endogenously expressed in the secretory acinar epithelium of prostate glandular cells, as well as within the proximal tubules of the kidney, salivary glandular cells, and the jejunal brush border of the small intestine to regulate folate metabolism[68-70]. PSMA also functions within nervous system glial cells, such as astrocytes and Schwann cells, to modulate glutamate signaling[71]. However, PSMA has also served as a therapeutic target TAA for prostate cancer due to its large extracellular domain, weak expression under normal physiologic conditions, and its profound degree of increased expression in prostate adenocarcinoma of 100 to
PSMA has already demonstrated efficacy as a radionucleotide target for imaging and as a therapeutic target for radioligand theranostic therapy. These developments have allowed for radiologic detection of PSMA-positive disease, including disease recurrence, which was otherwise not detected with conventional imaging modalities[73-75]. Theranostic treatment utilizing 177Lu-PSMA-617, a beta-emitting radionucleotide, has demonstrated antitumor response with associated radiographic response in patients with metastatic castrate-resistant prostate cancer. In the landmark phase III randomized control VISION trial, 831 patients demonstrated remarkable improvements in progression-free survival (8.7 vs. 3.4 months) and overall survival (15.3 vs. 11.3 months) compared to standard-of-care treatment, which ultimately led to FDA approval of the therapy[74-77].
In addition, PSMA-directed antibody-drug conjugate (ADC) therapy has also emerged as a novel treatment strategy. ADCs are composed of monoclonal antibodies that target TAAs of interest and are chemically linked to a cytotoxic agent that is delivered upon binding of the antigen. ADCs targeting PSMA under development are primarily delivering antimicrotubule cytotoxic compounds including maytansinoid-1 (DM1) (NCT00052000, NCT00070837) and monomethyl auristatin E (MMAE) (NCT01414283, NCT01695044)[35]. The efficacy of PSMA targeting ADCs is currently being evaluated in phase I/II clinical trials, with further research necessary to evaluate their therapeutic potential.
PSMA is also an attractive target for immunotherapy. CAR-T therapy targeting PSMA initially gained interest when CAR-T preclinical studies demonstrated strong antitumor activity and a robust immunogenic response including increased T-cell proliferation, cytolysis of tumor cells, and cytokine production in
A second-generation anti-PSMA CAR-T therapy was studied in a phase I dose-escalation trial (NCT01140373) with 7 enrolled patients who were treated with three different dosages. Patients in higher dose cohorts experienced high-grade fevers that were associated with increased levels of inflammatory cytokines (IL-4, 6, 8, 10, sIL-2ra) indicative of T-cell activation[82]. Another phase I dose-escalation trial was conducted with the enrollment of 5 patients treated with a first-generation anti-PSMA CAR, and findings demonstrated a > 50% PSA decline in two out of five patients and a minor response in a third patient[83].
Several ongoing trials investigating the efficacy of anti-PSMA CAR-T therapy have demonstrated promising preliminary results. In particular, an open-label, multicenter phase I dose-escalation trial (NCT04249947) employing P-PSMA-101 CAR-T has demonstrated encouraging preliminary data. P-PSMA-101 is a CAR designed with the piggyBac DNA Modification System which preferentially increases the production of stem cell memory cells, leading to long-term immunoactivation with T-cell expansion and the potential to alter the TME. Preliminary data of 10 heavily pre-treated patients with mCRPC who had received, on average, seven lines of prior therapy demonstrated a PSA decline of > 50% in three patients and > 99% in one patient. Further, three of the four patients demonstrated a complete radiographic response on PSMA-PET imaging with a concurrent decline in circulating tumor cell (CTC) burden. Histologic examination of one case demonstrated P-PSMA-101 CAR-T cell infiltration and a complete pathologic response with resolution of metastatic disease on interval imaging and biopsy results. An encouraging safety profile was also reported with only grade 1-3 AEs[84]. Studies evaluating the efficacy of dnTGF-BRII are currently ongoing, but preliminary data have identified dose-limiting toxicities despite efficacy in some, including deep responses[35,85].
PSMA targeting BiTE therapy is also in development with several ongoing clinical trials. Pasotuxizumab (AMG 212 or BAY 2010112) was the first BiTE therapy in clinical study to target PSMA. In vitro study of the drug demonstrated high PSMA antigen specificity[86], which ultimately led to the launch of a phase I dose-escalation study (NCT01723475) to determine the safety and maximum tolerated dose (MTD). In this study, 47 patients were enrolled in either a subcutaneous (SC) delivery group (31 patients) or a continuous IV (cIV) group (16 patients).
Results of the trial were encouraging, with a PSA decline of > 50% in nine patients with SC dosing and three patients with cIV dosing - two of whom demonstrated a sustained and durable response. However, all 31 patients in the SC group developed neutralizing anti-drug antibodies and this arm of the study was discontinued due to non-viable drug formulation. The cIV treatment group demonstrated a PSA reduction in 14 out of 16 patients and a dose-dependent CTC reduction. All patients experienced at least one AE, with the most common symptoms being fever (85%), chills (38%), and fatigue (34%). Nevertheless, the trial was terminated prior to the identification of a MTD. The extremely short half-life of the drug required continuous infusion, which proved to be logistically challenging for clinical use[87].
Acapatamab (AMG 160), a second-generation BiTE, required a short IV infusion administered every 14 days and boasted an increased half-life compared to Pasotuxizumab. Preclinical studies suggested that therapy upregulated PD-L1 and elicited a strong T-cell activation signal. Phase I study (NCT03792841) of acapatamab enrolled 32 patients and treatment was associated with a PSA reduction in 63% of patients; however, 84.4% experienced AEs, with cytokine release syndrome being the most common. The trial was terminated without reaching a MTD[88,89]. A phase I/II (NCT04631601) trial is currently ongoing to evaluate the therapeutic potential of acapatamab in combination with either abiraterone, enzalutamide, or AMG 404, a PD-1 receptor monoclonal antibody[90].
AMG 340 is yet another PSMA targeting BiTE, which has been engineered with a lower CD3 affinity compared to prior BITE iterations in efforts to reduce OTOT toxicities. An ongoing phase I dose escalation trial (NCT04740034) is currently recruiting patients to evaluate the safety profile and MTD.
Several innovative BiTE designs are currently being engineered to attempt to mitigate AEs, prolong half-life, and improve the efficacy of previous-generation therapy. APVO4141 (MOR209 or ES414) was one such construct that demonstrated improved half-life and stability; however, treated patients experienced high levels of immune activation leading to systemic toxicity and the development of anti-drug antibodies, resulting in early termination of the drug[91]. Similarly, JNJ-081 (JNJ-63898081) was designed with the potential of increased drug stability and improved safety profile, yet phase I study (NCT03926013) demonstrated minimal PSA reductions, high incidence of treatment-related AEs (most commonly pyrexia and CRS), and anti-drug antibody formation that resulted in cessation of the trial[92]. HPN424 is another BiTE developed with a third binding domain to albumin, in the hopes of increasing half-life. Phase I/IIa testing (NCT03577028) of 80 patients with metastatic castrate-resistant prostate cancer demonstrated a PSA reduction in 21% of patients and a CTC decline in 57%. AEs were notable for grade 3 AST increase (18%), ALT increase (11%), and anemia (11%), with CRS occurring in 64% of patients but only 4% > grade 3. MTD was not achieved and HPN424 was deemed not suitable for further studies based on its treatment efficacy and risk/benefit profile[93]. Another novel BiTE, CC-1, has shown preliminary results indicative of prolonged half-life with tolerable AEs, including no CRS of > grade 2 events[94]. Phase I trial of CC-1 is currently recruiting patients (NCT04104607). Furthermore, BiTE therapy targeting both PSMA and KLK2 (Kallikrein-Related Peptidase 2) TAAs are currently recruiting patients in highly anticipated clinical studies (NCT06095089). Additional ongoing trials are summarized in Table 2.
Immuno-oncology clinical trials in prostate cancer
| Trial ID | Phase | Target | Drug | Indication | Primary endpoint |
| NCT00836654 | II/III | EpCAM (BiTE) | Catumaxomab (removab) | Malignant ascites + EpCAM-positive tumors | DLT, AE profile, ORR |
| NCT00635596 | I | EpCAM (BiTE) | Solitomab (AMG110/MT110) | Relapsed/refractory solid tumors | DLT, AE profile |
| NCT01723475 | I | PSMA (BiTE) | Pasotuxizumab (AMG212/BAY2010 112) | mCRPC | MTD, DLT, AE profile |
| NCT03792841 | I | PSMA (BiTE) | Acapatamab (AMG160 (HLE)) | mCRPC | DLT, AE profile |
| NCT04631601 | I/II | PSMA (BiTE) | Acapatamab (AMG160 (HLE)) | mCRPC | DLT, AE profile |
| NCT02262910 | I | PSMA (BiTE) | ES414 (MOR209/APVO411) | mCRPC | MTD, DLT |
| NCT03577028 | I | PSMA (BiTE) | HPN424 | mCRPC | DLT, ORR |
| NCT03926013 | I | PSMA (BiTE) | JNJ-63898081 (JNJ-081) | mCRPC | DLT, AE profile |
| NCT05441501 | I | PSMA (BiTE) | JNJ-80038114 | mCRPC | DLT, AE profile |
| NCT04104607 | I | PSMA (BiTE) | CC-1 | mCRPC | AE profile |
| NCT04077021 | I | PSMA (BiTE) | CCW702 | mCRPC | DLT, AE profile, ORR |
| NCT05125016 | I/II | PSMA (BiTE) | REGN4336 | mCRPC | DLT, AE profile, ORR |
| NCT03972657 | I/II | PSMA (BiTE) | REGN5678 | mCRPC, clear cell RCC | MTD, DLT, AE profile, ORR |
| NCT04740034 | I | PSMA (BiTE) | AMG340 (TNB585) | mCRPC | MTD, DLT, AE profile |
| NCT06095089 | I | PSMA/KLK2 (BiTE) | JNJ-87189401 + JNJ-78278343 | mCRPC | DLT, AE profile, ADA development, ORR, |
| NCT05369000 | I/IIa | PSMA (gammabody BiTE) | LAVA-1207 | mCRPC | DLT, AE profile |
| NCT04839991 | I | PSMA (Trispecific T-cell enhancer BiTE) | CB307 | mCRPC | MTD, DLT, AE profile |
| NCT04227275 | I/IIa | PSMA (CAR-T) | CAR-T PSMA-TGF RDN cells | mCRPC | DLT, AE profile, ORR |
| NCT01140373 | I | PSMA (CAR-T) | Autologous anti- PSMA CAR-T cells | mCRPC | DLT, AE profile |
| NCT03089203 | I | PSMA (CAR-T) | CAR-T PSMA-TGF RDN cells | mCRPC | AE profile, T-cell expansion |
| NCT04249947 | I | PSMA (CAR-T) | P-PSMA-101 CAR-T cells | mCRPC | DLT, AE profile, ORR |
| NCT04429451 | I/II | PSMA (CAR-T) | 4SCAR-PSMA T-cells | PSMA positive tumors | DLT, AE profile |
| NCT04633148 | I | PSMA (CAR-T) | UniCAR02 T-cells | mCRPC | MTD, DLT, AE profile |
| NCT04768608 | I | PSMA/PD1 (CAR-T) | PD1-PSMA CAR-T cells | mCRPC | DLT, AE profile |
| NCT05354375 | I | PSMA (CAR-T) | Anti-PSMA CAR-T cells | mCRPC | DLT, AE profile, ORR |
| NCT05489991 | I/II | PSMA (CAR-T) | TmPSMA-02 (dual armored CAR-T cells) | mCRPC | DLT, AE profile, ORR |
| NCT05656573 | I | PSMA (CAR-T) | Anti-PSMA CAR-T cells | mCRPC | DLT, AE profile |
| NCT03692663 | I | PSMA (CAR- NK) | Anti-PSMA CAR NK cells (TABP EIC) | mCRPC | DLT, AE profile |
| NCT04053062 | I | PSMA (CAR-T) | LIGHT-PSMA-CAR-T | mCRPC | AE profile |
| NCT03927573 | I | PSCA (BITE) | GEM3PSCA | Prostate cancer, NSCLC, renal cancer | MTD, DLT, AE profile |
| NCT03873805 | I | PSCA (CAR-T) | Autologous anti- PSCA-CAR4-1BB/TCRzeta- CD19t-expressing T- lymphocytes | mCRPC | DLT, AE profile |
| NCT05805371 | I | PSCA (CAR-T) | Autologous anti- PSCA-CAR-4-1BB/TCRzeta- CD19t-expressing T- lymphocytes | mCRPC | DLT, AE profile |
| NCT05732948 | I | PSCA/PSMA/P D-1 (CAR-T) | Anti-PD-1/silent PSMA/PSCA targeted T-cells | mCRPC | DLT, AE profile |
| NCT02744287 | I/II | PSCA (CAR-T) | BPX-601 | mCRPC | MTD, DLT, AE profile |
| NCT04432649 | I/II | B7-H3 (CAR-T) | 4SCAR-276 | Recurrent/Refractory B7-H3-positive solid tumors | AE profile |
| NCT05515185 | I | B7-H3 (CAR-T) | Anti-B7-H3 CAR-T cells | Recurrent/Refractory B7-H3-positive solid tumors | Unknown |
| NCT04221542 | I | STEAP1 (BiTE) | AMG509 | mCRPC | DLT, AE profile |
| NCT04702737 | Ib | DLL3 (BiTE) | Tarlatamab (AMG757 (HLE)) | NEPC | DLT, AE profile, ORR |
Prostate stem cell antigen
Prostate stem cell antigen (PSCA) is a surface glycoprotein that was first identified as a TAA overexpressed in prostate cancer. PSCA has been hypothesized to be involved in intracellular signaling; however, its true physiologic function and regulatory mechanisms remain largely unknown[95]. PSCA has been noted to have minimal expression in non-neoplastic tissue, with its highest degree of expression in prostate glandular tissue; however, it is also present with limited expression in the bladder, pancreas, stomach, kidney, skin, and esophagus[95-97]. PSCA is expressed in 94% of primary prostate cancers and 100% of metastatic prostate cancers with bone involvement express PSCA[98], and increased PSCA expression correlates with increased Gleason grades and has been associated with advanced disease and poor prognosis[95-100].
Due to its expression profile, PSCA is an attractive TAA target for novel immunotherapy. Several ongoing, early-phase clinical trials are currently underway to explore BiTE and CAR-T therapy aimed at PSCA. Three ongoing phase I clinical trials are currently investigating the safety and tolerability profile of autologous anti-PSCA CAR-T drug delivery (NCT03873805, NCT05805371) and the combination of PD-1 silent PSMA/PSCA targeted CAR-T therapy (NCT05732948). BPX-601 (NCT02744287) was a PSCA CAR-T investigated in a phase I trial that was recently terminated due to dose-limiting toxicities. There are currently no active PSCA BiTE studies; however, a recent phase I trial of GEM3PSCA (NCT03927573), a PSCA × CD3 BiTE therapy, was terminated due to manufacturing challenges.
Six-transmembrane epithelial antigen of the prostate-1
Six-transmembrane epithelial antigen of the prostate-1 (STEAP-1) is a member of the STEAP1-4 protein family. STEAP proteins are metalloproteinases that regulate iron and copper homeostasis, relieve oxidative stress, and mediate the transferrin cycle[101-103]. STEAP proteins also serve an important role in apoptosis and cellular proliferation[104]. STEAP-1 is expressed at the surface of cell junctions in secretory prostate epithelial cells[101,102,104,105]. Similar to other TAAs of interest, STEAP-1 is overexpressed in prostate cancer with an estimated expression of > 80% and little to no expression in non-neoplastic tissues[106]. Trace expression of STEAP1 can be found in the bladder, ovaries, and bone marrow[74]. When STEAP-1 is overexpressed, tumor growth is promoted through unclear mechanisms. Hypothesized mechanisms suggest that during periods of overexpression, cell-cell junction connexins are inhibited and intracellular communication is mediated via STEAP1, which subsequently promotes stromal cell recruitment and malignant cell proliferation[101,104,106]. However, numerous studies have demonstrated an immunogenic role of STEAP-1 and its antitumor activity may be secondary to its effects on the immune milieu within the TME[74,101,106].
STEAP1 has become a focus of T-cell immunomodulatory drug development, with novel CAR-T and BiTE constructs actively being engineered[107-109]. AMG 509 is a BiTE therapy composed of two identical anti-STEAP-1 domains that have shown promising in vivo results with strong antitumor activity through increased cytotoxic T-cell activity and limited OTOT toxicity[110]. AMG 509 is currently being evaluated in the highly anticipated multicenter, open-label phase I study (NCT04221542) assessing the safety profile, pharmacokinetics, and efficacy as monotherapy and in conjunction with enzalutamide or abiraterone therapy[111]. This ongoing study has thus far enrolled 97 patients who have been treated with various dose levels of AMG 509. Early results have demonstrated that 15 patients (22.7%) had confirmed partial responses and 30 (45.5%) had stable disease. Declines in PSA levels of 50% and 90% occurred in 42 (47%) and 24 (27%), respectively. The most common adverse events were cytokine-release syndrome, mainly grade 1 or 2 in 72%, fatigue in 53%, anemia in 45%, fever in 40%, and myalgia in 405[112].
Additionally, STEAP-1 is the target for the development of numerous cancer vaccines. An mRNA vaccine was studied in a phase I/II trial (NCT01817738) that was terminated after data failed to demonstrate any signal in survival[101,113,114].
Tumor-associated antigens for neuroendocrine prostate cancer
Neuroendocrine prostate cancer (NEPC) is a poorly differentiated neuroendocrine carcinoma that broadly encompasses pure small cell, large cell, or mixed neuroendocrine carcinomas[115]. NEPC rarely occurs de novo and comprises less than 2% of all prostate cancers. However, upwards of 10%-15% of patients with metastatic castrate-resistant prostate cancer have been noted to develop treatment-related NEPC (t-NEPC) following treatment with androgen signaling inhibitors[116]. Evidence suggests that the development of t-NEPC may occur secondary to the inactivation of TP53 and RB1, which serve as epigenetic regulators. Diagnosis is often challenging and delayed as it typically presents in patients who have been diagnosed with advanced disease and are undergoing treatment directed at prostatic adenocarcinoma. Current treatment guidelines recommend first-line platinum-based chemotherapy regimens, but no standard treatment guidelines exist for second-line therapy and beyond[28,74,117]. Recent studies have demonstrated high expression of delta-like protein 3 (DLL3), a notch ligand, in NEPC disease. DLL3 was initially discovered due to overexpression of the antigen in small cell lung cancer (SCLC) surface epithelial cells[118-119], but DLL3 antigen expression has recently been described in 76.6% of evaluated NEPC cases. As such, DLL3 has emerged as a possible TAA target for immunotherapy. Tarlatamab (AMG 757) is a BiTE therapy currently under investigation in the phase Ib setting (NCT04702737) for the treatment of de novo or t-NEPC. There is also a CAR-T targeting DLL3 under investigation in SCLC (AMG 119) and it may have a potential application to NEPC depending on study results[74,117,120].
Recent studies have identified cell adhesion molecule 5, CEA (CEACAM5), as another highly expressed antigen specific to NEPC, and CEACAM5 may serve as a target TAA for future drug development. In vitro studies have demonstrated the successful creation of anti-CEA CAR-T cells with tumor growth delay and eradication[74,121,122]. While immunotherapy targeting the treatment of NEPC is in its early stages, there is an urgent and unmet need for novel therapies in this histology and future immunotherapy studies are likely to be forthcoming.
Alternative immunomodulatory targets for T-cell redirecting therapy
B7-H3 (CD276)
B7-H3 is a surface membrane protein that is a newly identified member of the B7 immunomodulator protein family. More well-known member proteins include PD-L1, PD-1, and CTLA-4[123-125]. While originally hypothesized to promote costimulatory immune activation, recent data suggest a dual role, with both costimulatory and, more predominantly, coinhibitory role for the antigen[125-129]. In addition, emerging studies have demonstrated that higher levels of B7-H3 have been associated with increased tumor survival, metastasis, and resistance to chemotherapy[125-130]. Furthermore, elevated levels of the antigen have been associated with higher Gleason scores, development of castrate-resistant metastatic disease, and poor overall prognosis[131]. Nonetheless, much is yet to be discovered regarding B7-H3’s immunologic and functional role within the TME. Similar to other TAAs of interest, B7-H3 has limited expression in normal tissue, with increased expression detected in solid tumor malignancies. Moreover, B7-H3 has been noted to overexpressed in greater than 80% of metastatic castrate-resistant prostate cancer cases, with overexpression also noted in CNS tumors (glioblastoma, neuroblastoma), ovarian, pancreatic, breast, colorectal, renal cell, non- small cell lung, and cutaneous squamous cell carcinoma[125,128-130].
Preclinical work targeting B7-H3 has demonstrated promising antitumor activity[125,127,130-134]. Other studies have demonstrated that defects in tumor suppressor genes, namely TP53 and PTEN, have been associated with increased B7-H3 levels. Other preclinical studies have observed that monoclonal antibodies, such as enoblituzumab, have been combined with PD-L1 inhibitors with dramatic success and have spurred further combination treatment approaches with ipilimumab, a CTLA-4 inhibitor, with clinical studies currently underway in patients with melanoma and non-small cell lung cancer (NCT02381314)[132-133]. Further drug development targeting B7-H3 is rapidly underway, encompassing multiple classes of therapy including ADCs (DS- 7300/MGC018), checkpoint inhibitors, tri-specific killer engager agents (TriKE), CAR natural killer cell agents (CAR-NK), and CAR-T therapies[126,127,132,133]. Topline results from early-phase studies of these therapies are demonstrating a favorable safety profile[127]. Studies employing B7-H3 targeting CAR-T therapies are ongoing (NCT04432649, NCT05515185).
PERSPECTIVES & FUTURE DIRECTIONS
Immunotherapy in prostate cancer represents an emerging treatment modality with promising therapeutic efficacy. Thus far, ICI has demonstrated only modest treatment responses; however, T-cell redirecting therapies are in the early stages of development and current progress should not be overlooked. The identification of multiple TAAs within neoplastic prostatic tissue has allowed for rapid developments in immuno-oncology. Nevertheless, barriers to the effective implementation of immunotherapy for patients with prostate cancer remain. Novel drug design targeting an appropriate TAA has the potential to overcome cancer immune evasion, which is a primary obstacle hindering immunotherapy in prostate cancer. Through immune evasion, tumors can escape immune-mediated elimination through three key mechanisms: (1) loss of antigenicity; (2) loss of immunogenicity; and (3) an immunosuppressive tumor microenvironment[135,136]. MHC class I downregulation is common in advanced prostate cancer and contributes to the loss of antigenicity and prevents subsequent immune-mediated mechanisms of cancer elimination[137,138]. Further, tumor cells have demonstrated the ability to secrete immunosuppressive cytokines, recruit regulatory T-cell subsets, and suppress antitumor effects that result in the alteration of the TME and create a “cold” immunosuppressive environment[135,139,140]. Implementation of ICIs in prostate cancer had little impact on altering the immune milieu of the TME. While newly engineered BiTE and CAR-T therapies hold promise in overcoming the multiple barriers created by immune evasion mechanisms, multimodal combination therapy across multiple classes of immunotherapy has the potential to synergistically target malignant cells while also converting the TME from a “cold” to “hot”. Additional studies with well-designed correlative analyses are required to further characterize immune escape mechanisms within the TME with respect to multimodality combination approaches.
Treatment-related toxicity with T-cell redirection therapy, namely CRS and OTOT, has resulted in the early termination of several clinical trials. Despite early concerns, studies have suggested that the administration of steroid and tocilizumab does not blunt T-cell response or negatively affect treatment efficacy, and that early intervention on toxicity may optimally prevent morbidity and allow for maximal benefit from therapy[141,142].
OTOT toxicity has decreased with the advent of highly specific TAA selection and with newly proposed mechanisms to mitigate this toxicity, one of which is decreased scFvs affinity for the TAA, as high specificity has been shown to lead to increased tumor effects but also increased OTOT effects. Novel strategies requiring further investigation include multistep activation parameters to ensure the drug is present within the TME, such as selective activation within an acidic pH or hypoxic tumor environment, or switches - permanent or reversible - that allow for inactivation of the drug as a safety mechanism to avoid severe AEs. Alternative strategies include regional administration of therapies, rather than systemic administration, in an attempt to concentrate antitumor activity to the TME without undesired systemic toxicity[47].
BiTE therapy initially faced logistical challenges due to a short half-life which necessitated continuous IV administration, while subcutaneous administration of the drug resulted in the formation of anti-drug antibodies, rendering this therapeutic class clinically impractical.
Fortunately, newer-generation BiTEs have optimized drug delivery with half-life-extended (HLE) formulations. While BiTE therapy boasts the potential of “off-the-shelf” manufacturing, allowing for cost-effective drug production without the cost or time needed for individual customization, further studies are necessary to identify appropriate TAAs in hopes of restricting target effect further improving safety profiles[26,28,35,74,143,144]. CAR-T therapy has faced similar hurdles, but specific limitations include the necessity to individually tailor therapy for patients, which is an expensive and time-consuming process, ultimately limiting availability. The need for prior lymphodepletion also poses a significant risk of infection and immunosuppression in a high-risk patient population[26,74,145,146]. Furthermore, traditional CAR-T delivery is not compatible with future off-the-shelf drug manufacturing. Both classes of T-cell redirecting therapies face the challenge of acquired treatment resistance due to TAA downregulation as an immune evasion mechanism. Combination therapy with ICIs and targeting multiple TAAs simultaneously have been proposed as strategies to overcome this barrier[26,28,35,74]. Moreover, the clinical data at hand surrounding T-cell redirecting therapies come from studies conducted in heavily pre-treated castration-resistant patient populations, who typically harbor an exhausted T-cell phenotype with a predominance of T-regulatory cell subsets within the TME[143,147,148]. As such, trial design with implementation of these therapies earlier in the natural history of the disease may prove the novel immunotherapies to be more efficacious than current data suggests.
The strong interest in prostate cancer immune-oncology has spurred the creation of several new drug constructs, including dual affinity retargeting bispecific antibodies (DART), simultaneous multiple interaction T-cell engagers (SMITE), CAR-NK, bispecific killer engagers (BiKE), and tri-specific killer engager agents (TriKE). DART and SMITE therapy are extensions of BiTE therapy that attempt to simultaneously target multiple TAAs in order to avoid acquired treatment resistance that is the experience commonly seen with BiTE administration in hematologic malignancies[26,66]. Alternatively, CAR-NK therapy is similar to CAR-T therapy; however, this treatment modality functions by proliferation and activation of NK cells rather than T-cells. In preclinical models, CAR-NK cells demonstrate the ability to retain their innate ability to identify target cells with downregulated TAAs, while also demonstrating targeted tumor cellular lysis with dual target antigen domains. NK cells also have a shorter life span compared to T- cells, which is suspected to decrease drug-related toxicities. Furthermore, this construct boasts the possibility of off-the-shelf manufacturing, as drug creation does not require individual customization[66,149]. BiKE therapy is mechanistically similar to BiTE therapy, but the anti-CD3 domain is replaced by anti-CD16 to target NK cell activation instead of T-cells. TriKE follow the same mechanistic principles but theoretically further enhance NK cell activation and function[66,149-151]. Additionally, cancer vaccine development has undergone a new resurgence with the employment of novel TAAs and new vaccine formulations that aim to function with both humoral and adaptive immunity with the intention of antigen spread and possible synergistic therapeutic efficacy with ICI administration[152]. Antigen-drug conjugate (ADC) therapies are rapidly emerging agents composed of a monoclonal antibody with a covalently linked cytotoxic load, garnering increasing attention in immunotherapy in recent times. ADCs are actively being designed to target many of the discussed TAAs, as well as other antigen targets, with promising preliminary results[153]. Additional antigen targets with encouraging immunotherapy advancements undergoing active study and development are referenced in Table 3.
Additional immunotherapy antigen targets not restricted to T-cell engager therapy
CONCLUSION
Immunotherapy in prostate cancer offers an opportunity for innovative treatment approaches with encouraging therapeutic potential. T-cell redirecting therapies have shown robust antitumor activity correlating with immune activation in preclinical and early-phase clinical trials. The therapeutic potential of these therapies relies on the emergence of TAAs for which next-generation BiTE and CAR-T constructs have been designed and are under investigation, with preliminary data suggesting improved safety profiles and increased efficacy. Furthermore, a multitargeted approach aimed at several TAAs may be the key to enhancing immunogenicity and obtaining durable responses and also represents a promising synergistic strategy to alter the “cold” immune milieu of prostate cancer. There is optimism that ongoing and future studies directing immunotherapy towards novel TAAs will dramatically alter the treatment landscape and revolutionize the treatment of prostate cancer.
DECLARATIONS
Author’s contribution
Conception and design: Bhasin A, Zarrabi KK
Manuscript writing: Bhasin A, Mille PJ, Eturi A, Iskander A, Tester W, Zarrabi KK
Availability of data and material
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. American cancer society. Facts & figures 2023. American Cancer Society. Atlanta, CA, USA. 2023. Available from: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2023-cancer-facts-figures.html [Last accessed on 4 Feb 2024].
2. National cancer institute. Cancer stat facts: prostate cancer. Available from: https://seer.cancer.gov/statfacts/html/prost.html [Last accessed on 4 Feb 2024].
3. Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin 1972;22:232-40.
4. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001;1:34-45.
5. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of "tumor escape" phenotypes. Nat Immunol 2002;3:999-1005.
6. Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005;8:369-80.
7. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23.
8. Motzer RJ, Escudier B, McDermott DF, et al. CheckMate 025 Investigators. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803-13.
9. Schmid P, Adams S, Rugo HS, et al. IMpassion130 Trial Investigators. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 2018;379:2108-21.
10. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. KEYNOTE-189 Investigators. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med 2018;378:2078-92.
11. Kantoff PW, Higano CS, Shore ND, et al. IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411-22.
12. Tucker MD, Zhu J, Marin D, et al. Pembrolizumab in men with heavily treated metastatic castrate-resistant prostate cancer. Cancer Med 2019;8:4644-55.
13. Subudhi SK, Siddiqui BA, Aparicio AM, et al. Combined CTLA-4 and PD-L1 blockade in patients with chemotherapy-naïve metastatic castration-resistant prostate cancer is associated with increased myeloid and neutrophil immune subsets in the bone microenvironment. J Immunother Cancer 2021;9:e002919.
14. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54.
15. Petrylak DP, Loriot Y, Shaffer DR, et al. Safety and clinical activity of atezolizumab in patients with metastatic castration-resistant prostate cancer: a phase I study. Clin Cancer Res 2021;27:3360-9.
16. Powles T, Yuen KC, Gillessen S, et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: a randomized phase 3 trial. Nat Med 2022;28:144-53.
17. Agarwal N, McGregor B, Maughan BL, et al. Cabozantinib in combination with atezolizumab in patients with metastatic castration-resistant prostate cancer: results from an expansion cohort of a multicentre, open-label, phase 1b trial (COSMIC-021). Lancet Oncol 2022;23:899-909.
18. Karzai F, VanderWeele D, Madan RA, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer 2018;6:141.
19. Hotte S, Winquist E, Chi K, et al. CCTG IND 232: A phase II study of durvalumab with or without tremelimumab in patients with metastatic castration resistant prostate cancer (mCRPC). Ann Oncol 2019;30:v885.
20. Antonarakis ES, Piulats JM, Gross-Goupil M, et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II keynote-199 study. J Clin Oncol 2020;38:395-405.
21. Graff JN, Tagawa ST, Hoimes CJ, et al. Pembrolizumab plus enzalutamide for enzalutamide-resistant metastatic castration- resistant prostate cancer (mCRPC): updated analyses after one additional year of follow-up from cohorts 4 and 5 of the keynote-199 study. 2021;39:5042.
22. Yu EY, Piulats JM, Gravis G, et al. Pembrolizumab plus olaparib in patients with metastatic castration-resistant prostate cancer: long-term results from the phase 1b/2 keynote-365 cohort a study. Eur Urol 2023;83:15-26.
23. Beer TM, Kwon ED, Drake CG, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 2017;35:40-7.
24. Kwon ED, Drake CG, Scher HI, et al. CA184-043 Investigators. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 2014;15:700-12.
25. Shenderov E, Boudadi K, Fu W, et al. Nivolumab plus ipilimumab, with or without enzalutamide, in AR-V7-expressing metastatic castration-resistant prostate cancer: a phase-2 nonrandomized clinical trial. Prostate 2021;81:326-38.
26. Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol 2020;17:418-34.
27. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol 2015;93:290-6.
28. Simão DC, Zarrabi KK, Mendes JL, et al. Bispecific t-cell engagers therapies in solid tumors: focusing on prostate cancer. Cancers 2023;15:1412.
30. Einsele H, Borghaei H, Orlowski RZ, et al. The BiTE (bispecific T-cell engager) platform: development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer 2020;126:3192-201.
32. Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol 2015;35 Suppl:S185-98.
33. Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol 2006;43:763-71.
34. Ross SL, Sherman M, McElroy PL, et al. Bispecific T cell engager (BiTE®) antibody constructs can mediate bystander tumor cell killing. PLoS One 2017;12:e0183390.
35. Zarrabi KK, Narayan V, Mille PJ, et al. Bispecific PSMA antibodies and CAR-T in metastatic castration-resistant prostate cancer. Ther Adv Urol 2023;15:17562872231182219.
36. Aldoss I, Bargou RC, Nagorsen D, Friberg GR, Baeuerle PA, Forman SJ. Redirecting T cells to eradicate B-cell acute lymphoblastic leukemia: bispecific T-cell engagers and chimeric antigen receptors. Leukemia 2017;31:777-87.
37. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 1989;86:10024-8.
38. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018;359:1361-5.
39. Wrzesinski C, Paulos CM, Kaiser A, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother 2010;33:1-7.
40. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci 2019;20:1283.
41. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014;124:188-95.
42. Van De Vyver AJ, Marrer-Berger E, Wang K, Lehr T, Walz AC. Cytokine release syndrome by T-cell-redirecting therapies: can we predict and modulate patient risk? Clin Cancer Res 2021;27:6083-94.
43. Maus MV, Alexander S, Bishop MR, et al. Society for immunotherapy of cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer 2020;8:e001511.
44. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol 2022;22:85-96.
45. Klinger M, Zugmaier G, Nägele V, et al. Adhesion of T cells to endothelial cells facilitates blinatumomab-associated neurologic adverse events. Cancer Res 2020;80:91-101.
46. Stein AS, Schiller G, Benjamin R, et al. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: management and mitigating factors. Ann Hematol 2019;98:159-67.
47. Flugel CL, Majzner RG, Krenciute G, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol 2023;20:49-62.
48. Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans 2016;44:951-9.
49. Guo Y, Feng K, Liu Y, et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res 2018;24:1277-86.
50. Feng K, Liu Y, Guo Y, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018;9:838-47.
51. Thistlethwaite FC, Gilham DE, Guest RD, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother 2017;66:1425-36.
53. Gunia S, Koch S, May M, Dietel M, Erbersdobler A. Expression of prostatic acid phosphatase (PSAP) in transurethral resection specimens of the prostate is predictive of histopathologic tumor stage in subsequent radical prostatectomies. Virchows Arch 2009;454:573-9.
54. Schaeffer EM, Srinivas S, Adra N, et al. NCCN guidelines® insights: prostate cancer, version 1.2023. J Natl Compr Canc Netw 2022;20:1288-98.
55. Madan RA, Antonarakis ES, Drake CG, et al. Putting the pieces together: completing the mechanism of action jigsaw for sipuleucel-T. J Natl Cancer Inst 2020;112:562-73.
56. Cheng ML, Fong L. Beyond sipuleucel-T: immune approaches to treating prostate cancer. Curr Treat Options Oncol 2014;15:115-26.
57. Schnell U, Cirulli V, Giepmans BN. EpCAM: structure and function in health and disease. Biochim Biophys Acta 2013;1828:1989-2001.
58. Balzar M, Winter MJ, de Boer CJ, Litvinov SV. The biology of the 17-1A antigen (Ep-CAM). J Mol Med 1999;77:699-712.
59. Huang L, Yang Y, Yang F, et al. Functions of EpCAM in physiological processes and diseases (review). Int J Mol Med 2018;42:1771-85.
60. Zhou N, Wang H, Liu H, et al. MTA1-upregulated EpCAM is associated with metastatic behaviors and poor prognosis in lung cancer. J Exp Clin Cancer Res 2015;34:157.
61. Massoner P, Thomm T, Mack B, et al. EpCAM is overexpressed in local and metastatic prostate cancer, suppressed by chemotherapy and modulated by MET-associated miRNA-200c/205. Br J Cancer 2014;111:955-64.
62. Heiss MM, Murawa P, Koralewski P, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: results of a prospective randomized phase II/III trial. Int J Cancer 2010;127:2209-21.
63. Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs 2010;2:129-36.
64. Mau-Sørensen M, Dittrich C, Dienstmann R, et al. A phase I trial of intravenous catumaxomab: a bispecific monoclonal antibody targeting EpCAM and the T cell coreceptor CD3. Cancer Chemother Pharmacol 2015;75:1065-73.
65. Kebenko M, Goebeler ME, Wolf M, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology 2018;7:e1450710.
66. Zorko NA, Ryan CJ. Novel immune engagers and cellular therapies for metastatic castration-resistant prostate cancer: do we take a BiTe or ride BiKEs, TriKEs, and CARs? Prostate Cancer Prostatic Dis 2021;24:986-96.
67. Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res 1997;3:81-5.
68. Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc Natl Acad Sci USA 2005;102:5981-6.
69. Mhawech-Fauceglia P, Zhang S, Terracciano L, et al. Prostate-specific membrane antigen (PSMA) protein expression in normal and neoplastic tissues and its sensitivity and specificity in prostate adenocarcinoma: an immunohistochemical study using mutiple tumour tissue microarray technique. Histopathology 2007;50:472-83.
70. Kinoshita Y, Kuratsukuri K, Landas S, et al. Expression of prostate-specific membrane antigen in normal and malignant human tissues. World J Surg 2006;30:628-36.
71. Kaittanis C, Andreou C, Hieronymus H, et al. Prostate-specific membrane antigen cleavage of vitamin B9 stimulates oncogenic signaling through metabotropic glutamate receptors. J Exp Med 2018;215:159-75.
72. Heston WD. Characterization and glutamyl preferring carboxypeptidase function of prostate specific membrane antigen: a novel folate hydrolase. Urology 1997;49:104-12.
73. Rahn KA, Slusher BS, Kaplin AI. Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition. Curr Med Chem 2012;19:1335-45.
74. Dorff TB, Narayan V, Forman SJ, et al. Novel redirected T-cell immunotherapies for advanced prostate cancer. Clin Cancer Res 2022;28:576-84.
75. O'Keefe DS, Bacich DJ, Huang SS, Heston WDW. A perspective on the evolving story of PSMA biology, PSMA-based imaging, and endoradiotherapeutic strategies. J Nucl Med 2018;59:1007-13.
76. Rowe SP, Campbell SP, Mana-Ay M, et al. Prospective evaluation of PSMA-targeted 18F-DCFPyL PET/CT in men with biochemical failure after radical prostatectomy for prostate cancer. J Nucl Med 2020;61:58-61.
77. Hofman MS, Emmett L, Sandhu S, et al. TheraP Trial Investigators and the Australian and New Zealand Urogenital and Prostate Cancer Trials Group. [177Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomised, open-label, phase 2 trial. Lancet 2021;397:797-804.
78. Hofman MS, Violet J, Hicks RJ, et al. [177Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): a single-centre, single-arm, phase 2 study. Lancet Oncol 2018;19:825-33.
79. Sartor O, de Bono J, Chi KN, et al. VISION Investigators. Lutetium-177-PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med 2021;385:1091-103.
80. Ma Q, Gomes EM, Lo AS, Junghans RP. Advanced generation anti-prostate specific membrane antigen designer T cells for prostate cancer immunotherapy. Prostate 2014;74:286-96.
81. Boyerinas B, Miller S, Murray R, et al. Abstract 602: a novel TGF-β/IL-12R signal conversion platform that protects CAR T cells from TGF-β-mediated immune suppression and concurrently amplifies effector function. Cancer Res 2017;77:602.
82. Calcinotto A, Spataro C, Zagato E, et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018;559:363-9.
83. Wang D, Shao Y, Zhang X, Lu G, Liu B. IL-23 and PSMA-targeted duo-CAR T cells in prostate cancer eradication in a preclinical model. J Transl Med 2020;18:23.
84. Slovin SF, Wang X, Hullingsand M, et al. Chimeric antigen receptor (CAR+) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). J Clin Oncol 2013;31:72.
85. Junghans RP, Ma Q, Rathore R, et al. Phase I trial of anti-psma designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate 2016;76:1257-70.
86. Slovin SF, Dorff TB, Falchookand GS, et al. Phase 1 study of P-PSMA-101 CAR-T cells in patients with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2022;40:98.
87. Narayan V, Barber-Rotenberg JS, Jung IY, et al. Prostate Cancer Cellular Therapy Program Investigators. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med 2022;28:724-34.
88. Friedrich M, Raum T, Lutterbuese R, et al. Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol Cancer Ther 2012;11:2664-73.
89. Hummel HD, Kufer P, Grüllich C, et al. Pasotuxizumab, a BiTE® immune therapy for castration-resistant prostate cancer: phase I, dose-escalation study findings. Immunotherapy 2021;13:125-41.
90. Deegen P, Thomas O, Nolan-Stevaux O, et al. The PSMA-targeting half-life extended BiTE therapy AMG 160 has potent antitumor activity in preclinical models of metastatic castration-resistant prostate cancer. Clin Cancer Res 2021;27:2928-37.
91. Tran B, Horvath L, Dorff T, et al. 609O results from a phase I study of AMG 160, a half-life extended (HLE), PSMA-targeted, bispecific T-cell engager (BiTE®) immune therapy for metastatic castration-resistant prostate cancer (mCRPC). Ann Oncol 2020;31:S507.
92. Subudhi SK, Siddiqui BA, Maly JJ, et al. Safety and efficacy of AMG 160, a half-life extended BiTE immune therapy targeting prostate-specific membrane antigen (PSMA), and other therapies for metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2021;39:TPS5088.
93. Hernandez-Hoyos G, Sewell T, Bader R, et al. MOR209/ES414, a novel bispecific antibody targeting PSMA for the treatment of metastatic castration-resistant prostate cancer. Mol Cancer Ther 2016;15:2155-65.
94. Lim EA, Schweizer MT, Chi KN, et al. Phase 1 study of safety and preliminary clinical activity of JNJ-63898081, a PSMA and CD3 bispecific antibody, for metastatic castration-resistant prostate cancer. Clin Genitourin Cancer 2023;21:366-75.
95. De Bono JSD, Fong L, Beer TM, et al. Results of an ongoing phase 1/2a dose escalation study of HPN424, a tri-specific half-life extended PSMA-targeting T-cell engager, in patients with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2021;39:5013.
96. Heitmann JS, Walz JS, Pflügler M, et al. Abstract CT141: CC-1, a bispecific PSMAxCD3 antibody for treatment of prostate carcinoma: results of the ongoing phase I dose escalation trial. Cancer Res 2022;82:CT141.
97. Reiter RE, Gu Z, Watabe T, et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci USA 1998;95:1735-40.
98. Raff AB, Gray A, Kast WM. Prostate stem cell antigen: a prospective therapeutic and diagnostic target. Cancer Lett 2009;277:126-32.
99. Saeki N, Gu J, Yoshida T, Wu X. Prostate stem cell antigen: a Jekyll and Hyde molecule? Clin Cancer Res 2010;16:3533-8.
100. Gu Z, Thomas G, Yamashiro J, et al. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000;19:1288-96.
101. Xu M, Evans L, Bizzaro CL, et al. STEAP1-4 (six-transmembrane epithelial antigen of the prostate 1-4) and their clinical implications for prostate cancer. Cancers 2022;14:4034.
102. Ohgami RS, Campagna DR, McDonald A, Fleming MD. The steap proteins are metalloreductases. Blood 2006;108:1388-94.
103. Knutson MD. Steap proteins: implications for iron and copper metabolism. Nutr Rev 2007;65:335-40.
104. Gomes IM, Rocha SM, Gaspar C, et al. Knockdown of STEAP1 inhibits cell growth and induces apoptosis in LNCaP prostate cancer cells counteracting the effect of androgens. Med Oncol 2018;35:40.
105. Yamamoto T, Tamura Y, Kobayashi J, et al. Six-transmembrane epithelial antigen of the prostate-1 plays a role for in vivo tumor growth via intercellular communication. Exp Cell Res 2013;319:2617-26.
106. Rodeberg DA, Nuss RA, Elsawa SF, Celis E. Recognition of six-transmembrane epithelial antigen of the prostate-expressing tumor cells by peptide antigen-induced cytotoxic T lymphocytes. Clin Cancer Res 2005;11:4545-52.
107. Lin TY, Park JA, Long A, Guo HF, Cheung NV. Novel potent anti-STEAP1 bispecific antibody to redirect T cells for cancer immunotherapy. J Immunother Cancer 2021;9:e003114.
108. Bhatia V, Kamat NV, Pariva TE, et al. Targeting advanced prostate cancer with STEAP1 chimeric antigen receptor T cell and tumor-localized IL-12 immunotherapy. Nat Commun 2023;14:2041.
109. Jin Y, Lorvik KB, Jin Y, et al. Development of STEAP1 targeting chimeric antigen receptor for adoptive cell therapy against cancer. Mol Ther Oncolytics 2022;26:189-206.
110. Li C, Fort M, Liang L, et al. 718 AMG 509, a STEAP1 × CD3 bispecific XmAb® 2+1 immune therapy, exhibits avidity-driven binding and preferential killing of high STEAP1-expressing prostate and Ewing sarcoma cancer cells. J Immunother Cancer 2020;8:A760.
111. Danila DC, Waterhouse DM, Appleman LJ, et al. A phase 1 study of AMG 509 in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2022;40:TPS5101.
112. Kelly WK, Danila DC, Lin CC, et al. Xaluritamig, a STEAP1 × CD3 XmAb 2+1 immune therapy for metastatic castration-resistant prostate cancer: results from dose exploration in a first-in-human study. Cancer Discov 2024;14:76-89.
113. Stenzl A, Feyerabend S, Syndikus I, et al. Results of the randomized, placebo-controlled phase I/IIB trial of CV9104, an mRNA based cancer immunotherapy, in patients with metastatic castration-resistant prostate cancer (mCRPC). J Immunother Cancer 2017;28:V408-9.
114. de la Garcia-Hernandez ML, Gray A, Hubby B, Kast WM. In vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: a candidate antigen for treating prostate cancer. Cancer Res 2007;67:1344-51.
115. Zaffuto E, Pompe R, Zanaty M, et al. Contemporary incidence and cancer control outcomes of primary neuroendocrine prostate cancer: a SEER database analysis. Clin Genitourin Cancer 2017;15:e793-800.
116. Bluemn EG, Coleman IM, Lucas JM, et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 2017;32:474-89.e6.
117. Yamada Y, Beltran H. Clinical and biological features of neuroendocrine prostate cancer. Curr Oncol Rep 2021;23:15.
118. Puca L, Gavyert K, Sailer V, et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci Transl Med 2019;11:eaav0891.
119. Hipp S, Voynov V, Drobits-Handl B, et al. A bispecific DLL3/CD3 IgG-like T-cell engaging antibody induces antitumor responses in small cell lung cancer. Clin Cancer Res 2020;26:5258-68.
120. Chou J, Egusa EA, Wang S, et al. Immunotherapeutic targeting and PET imaging of DLL3 in small-cell neuroendocrine prostate cancer. Cancer Res 2023;83:301-15.
121. Lee JK, Bangayan NJ, Chai T, et al. Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer. Proc Natl Acad Sci USA 2018;115:E4473-82.
122. Cha S, Yazaki P, Brown C, Shively J. Abstract PO083: treatment of CEA-positive solid tumors with anti-CEA chimeric antigen receptor T-cells in CEA transgenic mice. Cancer Immunol Res 2021;9:PO083.
123. Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res 2016;22:3425-31.
124. Li D, Xiang S, Shen J, et al. Comprehensive understanding of B7 family in gastric cancer: expression profile, association with clinicopathological parameters and downstream targets. Int J Biol Sci 2020;16:568-82.
125. Yang S, Wei W, Zhao Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int J Biol Sci 2020;16:1767-73.
126. Lanka SM, Zorko NA, Antonarakis ES, Barata PC. Metastatic castration-resistant prostate cancer, immune checkpoint inhibitors, and beyond. Curr Oncol 2023;30:4246-56.
127. Kontos F, Michelakos T, Kurokawa T, et al. B7-H3: an attractive target for antibody-based immunotherapy. Clin Cancer Res 2021;27:1227-35.
128. Guo C, Figueiredo I, Gurel B, et al. B7-H3 as a therapeutic target in advanced prostate cancer. Eur Urol 2023;83:224-38.
129. Shi X, Day A, Bergom HE, et al. Integrative molecular analyses define correlates of high B7-H3 expression in metastatic castrate-resistant prostate cancer. NPJ Precis Oncol 2022;6:80.
130. Li S, Zhang M, Wang M, et al. B7-H3 specific CAR-T cells exhibit potent activity against prostate cancer. Cell Death Discov 2023;9:147.
131. Benzon B, Zhao SG, Haffner MC, et al. Correlation of B7-H3 with androgen receptor, immune pathways and poor outcome in prostate cancer: an expression-based analysis. Prostate Cancer Prostatic Dis 2017;20:28-35.
132. Shi W, Wang Y, Zhao Y, et al. Immune checkpoint B7-H3 is a therapeutic vulnerability in prostate cancer harboring PTEN and TP53 deficiencies. Sci Transl Med 2023;15:eadf6724.
133. Mendes AA, Lu J, Kaur HB, et al. Association of B7-H3 expression with racial ancestry, immune cell density, and androgen receptor activation in prostate cancer. Cancer 2022;128:2269-80.
134. Shenderov E, De Marzo AM, Lotan TL, et al. Neoadjuvant enoblituzumab in localized prostate cancer: a single arm, phase 2 trial. Nat Med 2023;29:888-97.
135. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 2015;21:687-92.
136. Ferrone C, Dranoff G. Dual roles for immunity in gastrointestinal cancers. J Clin Oncol 2010;28:4045-51.
137. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011;331:1565-70.
138. Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene 2008;27:5869-85.
139. Le Mercier I, Chen W, Lines JL, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res 2014;74:1933-44.
140. Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med 2013;5:200ra116.
141. Gardner RA, Ceppi F, Rivers J, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood 2019;134:2149-58.
142. Liu S, Deng B, Yin Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J 2020;10:15.
143. Viardot A, Locatelli F, Stieglmaier J, Zaman F, Jabbour E. Concepts in immuno-oncology: tackling B cell malignancies with CD19-directed bispecific T cell engager therapies. Ann Hematol 2020;99:2215-29.
144. Li H, Er Saw P, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol 2020;17:451-61.
145. Perera MPJ, Thomas PB, Risbridger GP, et al. Chimeric antigen receptor T-cell therapy in metastatic castrate-resistant prostate cancer. Cancers 2022;14:503.
146. Braig F, Brandt A, Goebeler M, et al. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 2017;129:100-4.
147. Kamat NV, Yu EY, Lee JK. BiTE-ing into prostate cancer with bispecific T-cell engagers. Clin Cancer Res 2021;27:2675-7.
148. Correnti CE, Laszlo GS, de van der Schueren WJ, et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018;32:1239-43.
149. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006;107:159-66.
150. Schmohl JU, Felices M, Taras E, Miller JS, Vallera DA. Enhanced ADCC and NK cell activation of an anticarcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol Ther 2016;24:1312-22.
151. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. In: Somanchi SS, editor. Natural killer cells. New York: Springer; 2016. pp. 333-46.
152. Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer 2021;21:360-78.
153. Shastry M, Gupta A, Chandarlapaty S, Young M, Powles T, Hamilton E. Rise of antibody-drug conjugates: the present and future. Am Soc Clin Oncol Educ Book 2023;43:e390094.
154. Madan RA, Arlen PM, Mohebtash M, Hodge JW, Gulley JL. Prostvac-VF: a vector-based vaccine targeting PSA in prostate cancer. Expert Opin Investig Drugs 2009;18:1001-11.
155. Kyriakopoulos CE, Eickhoff JC, Ferrari AC, et al. Multicenter phase I trial of a DNA vaccine encoding the androgen receptor ligand-binding domain (pTVG-AR, MVI-118) in patients with metastatic prostate cancer. Clin Cancer Res 2020;26:5162-71.
156. Bardia A, Messersmith WA, Kio EA, et al. Sacituzumab govitecan, a trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol 2021;32:746-56.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Bhasin A, Mille PJ, Eturi A, Iskander A, Tester W, Zarrabi KK. Tumor-associated antigen targets for novel immune-based strategies in prostate cancer. J Transl Genet Genom 2024;8:55-76. http://dx.doi.org/10.20517/jtgg.2023.46
AMA Style
Bhasin A, Mille PJ, Eturi A, Iskander A, Tester W, Zarrabi KK. Tumor-associated antigen targets for novel immune-based strategies in prostate cancer. Journal of Translational Genetics and Genomics. 2024; 8(1): 55-76. http://dx.doi.org/10.20517/jtgg.2023.46
Chicago/Turabian Style
Bhasin, Amman, Patrick J. Mille, Aditya Eturi, Andrew Iskander, William Tester, Kevin K. Zarrabi. 2024. "Tumor-associated antigen targets for novel immune-based strategies in prostate cancer" Journal of Translational Genetics and Genomics. 8, no.1: 55-76. http://dx.doi.org/10.20517/jtgg.2023.46
ACS Style
Bhasin, A.; Mille PJ.; Eturi A.; Iskander A.; Tester W.; Zarrabi KK. Tumor-associated antigen targets for novel immune-based strategies in prostate cancer. J. Transl. Genet. Genom. 2024, 8, 55-76. http://dx.doi.org/10.20517/jtgg.2023.46
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 5
likes
Like This Article 5
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.