
Genomics and genetics of clear cell renal cell carcinoma: a mini-review

Abstract
Renal cell carcinoma (RCC) represents a heterogeneous group of malignancies derived from the kidney, of which clear cell RCC (ccRCC) accounts for nearly 75% of cases. Despite major advances in effective therapies, metastatic ccRCC is still associated with a 10%-20% 5-year survival and remains quite lethal. Great effort has been placed into understanding the genetics and genomics of ccRCC and their prognostic and therapeutic implications. Large-scale cancer genomics sequencing studies have identified several driver genes beyond VHL, particularly PBRM1 (40%), SETD2 (15%), and BAP1 (10%), drastically changing the concept of single-gene pathology underlying sporadic ccRCC. In this mini-review, we explore the pathways by which the loss of VHL, PBRM1, SETD2, and/or BAP1 induce ccRCC through discussion of gene function, disease models, prognostic indications, and therapeutic advances.
Keywords
Introduction
Renal cell carcinoma (RCC) represents a heterogeneous group of malignancies derived from the kidney, of which clear cell RCC (ccRCC) accounts for nearly 75% of cases[1]. ccRCC is characterized by lipid- and glycogen-rich cytoplasm, which appears clear following histologic processing. ccRCC has generally been defined by biallelic loss of the VHL tumor suppressor gene, located on 3p25 - loss of heterozygosity of 3p has been demonstrated in > 90% of ccRCC cases and complete loss of the VHL gene via genetic and/or epigenetic mechanisms have been shown in > 80% of cases[2]. Loss of VHL leads to uncontrolled activity of hypoxia-inducible transcription factors (HIFs), which promotes the inhibition of mitochondrion and redirection of glucose and glutamine for glycogen and lipid synthesis, leading to the classic morphological appearance of ccRCC[3].
However, due to large-scale cancer genomics sequencing efforts, several driver genes beyond VHL have been identified, and the concept of single-gene pathology underlying sporadic ccRCC has drastically evolved[4]. Prevalent gene mutations identified by these consortium studies included PBRM1 (40%), SETD2 (15%), and BAP1 (10%). These tumor suppressor genes are all located on 3p21 [Figure 1] and encode for chromatin and/or histone modifiers, suggesting epigenetic dysregulation as a convergent pathogenic signature in ccRCC[5,6]. Given the close genomic localization of all 4 genes spanning chromosome 3p21-25, the allelic loss of 3p is a fundamental component of ccRCC pathogenesis [Figure 1][2]. These pathways by which the loss of VHL, PBRM1, SETD2, and/or BAP1 induce disease will further be discussed in relation to function, disease models, prognostic indications, and therapeutic advances.
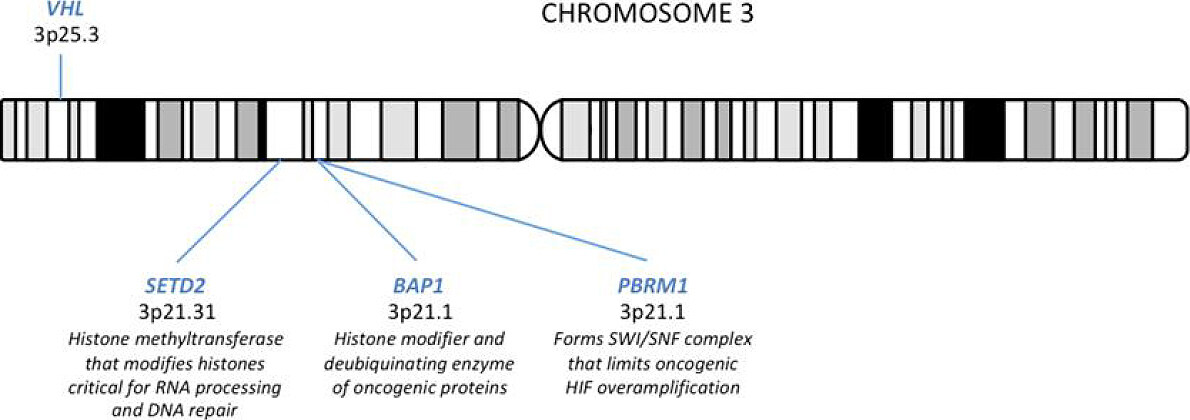
Figure 1. Chromosome 3p tumor suppressor genes of kidney cancer and their major functions
The VHL/HIF pathway
VHL
VHL plays a key role in the pathogenesis of ccRCC and its inactivation has been identified as the earliest and fundamental driving event in the development of ccRCC. The VHL tumor suppressor gene encodes for VHL, a component of the E3 ligase complex responsible for ubiquitination of hypoxia-inducible transcription factors 1α and 2α (HIF-1α and HIF-2α) for proteasome-mediated degradation[7,8]. In a normal oxygen state, HIF-1α and HIF-2α are modified by prolyl hydroxylase domain (PHD) proteins that allow VHL recognition and binding, leading to the rapid degradation of these HIF proteins[7,9]. Hypoxic conditions inactivate PHD proteins that perform the posttranslational prolyl hydroxylation of HIFs for degradation, thereby stabilizing HIF-1α and HIF-2α[7]. Similarly, loss of VHL function by somatic mutation, hypermethylation, or other genomic alterations of VHL, results in loss of HIF protein degradation and uninhibited HIF activity regardless of oxygen status. Recent genomic studies have also identified a group of ccRCC tumors that lack the characteristic 3p loss and VHL mutations but also display overexpression of HIF proteins[10]. These tumors are characterized by hotspot mutations in transcription elongation factor B (TCEB1), which encodes for protein elongin C, a subunit of the Elongin (SIII) complex that plays a critical role in eukaryotic transcription elongation[11]. This complex is crucial for the recruitment of VHL to the VCB (VHL-elongin C-elongin B) E3 ubiquitin ligase complex that is responsible for HIF degradation, leading to the unregulated HIF activity. Though TCEB1-mutated RCC tumors have yet to be established as a distinct subtype, identification of loss of TCEB1 and its effect on HIF activity supports the central HIF-driven pathogenic theme in ccRCC tumors. Furthermore, the activation of the HIF signaling pathway is also observed in several additional RCC subtypes, such as clear cell papillary RCC tumors which are morphologically and clinically distinct from ccRCC[12].
HIF
Upon stabilization, HIF-1α accumulates and directly regulates cellular metabolism in coping with the low oxygen environmental stress. Additionally, HIF-1α, like HIF-2α, dimerizes with HIF-1β or aryl hydrocarbon receptor nuclear translocator, producing an active transcription factor. HIF-1 and HIF-2 dimer complexes then activate and upregulate the transcription of numerous hypoxia-inducible targets genes, many of which mediate cell metabolism and growth[7]. These target genes differ among different cell lines and tissues, and the sets of genes regulated by HIF-1α and HIF-2α, respectively, overlap but are not identical[13]. In kidney cells, glycolysis is primarily driven by HIF-1α, whereas HIF-2α regulates expression of vascular endothelial growth factor A (VEGFA), cyclin D1, erythropoietin, and C-X-C chemokine receptor 4[14].
Increased HIF-1α levels have been associated with increased mortality in many types of human cancers[15]. However, the role of HIF-1 and HIF-2 in ccRCC has not yet been fully elucidated, as they seem to both affect the development and progression of ccRCC. Mouse models with constitutive HIF-1α, not HIF-2α, expression in the renal proximal tubule developed ccRCC[16,17]. Elimination of HIF-2α in VHL-/- ccRCC xenograft assays can suppress their ability to form tumors in nude mouse, while HIF-2α overproduction can override intact VHL tumor suppressor function[14,18,19]. Furthermore, HIF-2α single nucleotide polymorphisms have been associated with increased risk of developing kidney cancer in the general population[20]. Therefore in human ccRCC, disease progression and mortality have been associated with HIF-2α overexpression rather than HIF-1α, which is often silenced by gene deletion[21]. Altogether, HIF-1 probably plays an essential role in tumor initiation, whereas HIF-2 is needed for tumor progression.
Identification of HIFs as renal oncogenes and their downstream targets have led to development of many currently approved and investigational therapies for systemic treatment of advanced or locally unresectable ccRCC. Given the dependence of ccRCC tumors on angiogenesis for growth, inhibitors of VEGF and VEGF receptor tyrosine kinases, such as bevacizumab, sorafenib sunitinib, pazopanib, and axitinib, cabozantinib, and lenvatinib have been developed and are widely used as first or second-line therapies [Figure 2][22]. Small molecule inhibitors of HIF-2α have also been developed[23]. HIF-2α antagonism may theoretically provide additional efficacy than VEGF inhibitors[24]. Currently under investigation, PT2385, a direct HIF-2α inhibitor, has shown favorable safety profile and activity in patients with previously treated advanced ccRCC[9]. Additional approved and investigational treatments for ccRCC will be discussed in the “Current Therapies” section.
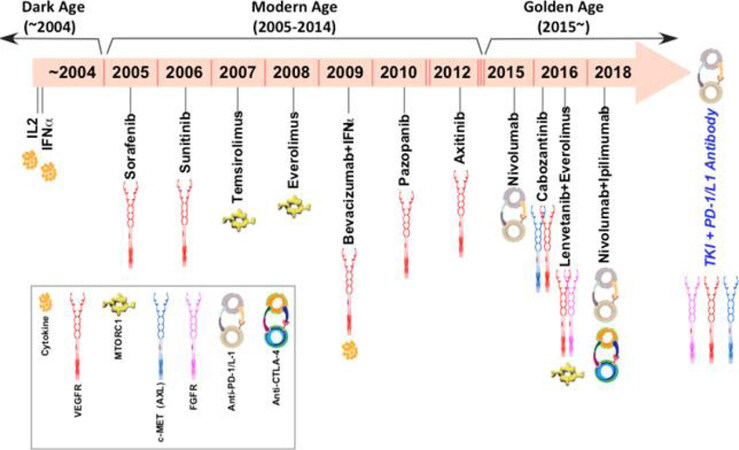
Figure 2. Diagram depicts the time line of renal cell carcinoma therapeutic development with 13 approved drugs representing 7 different mechanisms
Epigenetic and chromatin regulation pathways in ccRCC
Though it plays a key role in the pathogenesis of ccRCC, VHL loss alone is not sufficient for ccRCC development. Given the latency of ccRCC formation in human VHL syndrome and the inability to induce ccRCC in VHL-deficient mice[25], ccRCC development requires additional genetic and/or epigenetic events to occur. In order to characterize these driving events, several prevalent novel gene mutations have been identified, including PBRM1, SETD2, and BAP1. Located on 3p21, these genes encode for tumor suppressor chromatin- and histone- modifying proteins[26], and unlike VHL loss, PBRM1[27], SETD2[27], and BAP1[27,28] mutations are associated with more clinical progression of individual stages of ccRCC.
PBRM1
PBRM1 was first identified as the second most commonly mutated gene in ccRCC in 2010[29]. Confirmed in subsequent large-scale genomics studies, PBRM1 mutations occur in about 40% of human ccRCC cases[4,30]. PBRM1 encodes for protein BRG1-associated factor (BAF) 180, a critical subunit of the polybromo BAF SWI/SNF chromatin remodeling complex[30]. SWI/SNF chromatin remodeling complexes are large macromolecular structures that mobilize nucleosome via ATP consumption, thereby modulating chromatin structure to regulate vital cellular processes, including cell cycle, cell fate, cell death, metabolism, and DNA repair[30-32]. Detected in about 20% of human cancers[33], mutations in SWI/SNF proteins and other epigenetic regulators encompass a major class of cancer genes, demonstrating preferential enrichment by cancer type[34]. PBRM1, highly mutated in ccRCC, has been shown to play a critical role in the tumorigenesis of ccRCC.
Several studies have established the tumor suppressor role of PBRM1 via analysis of pre-neoplastic renal cells in kidney-specific PBRM1- and VHL-deficient mouse models and in RCC cell lines A704 and 786-O in vitro[30,35]. Mechanistically, genetically engineered mouse kidney cancer models demonstrated that PBRM1 functions to prevent kidney tumor cell growth by restraining the self-propagating over-amplification of HIF1 signaling by limiting the HIF1-STAT3 feed-forward loop[30,35].
In the same PBRM1-/- VHL-/- mouse model study, a long latency period of > 6 months was observed, again suggesting the involvement of an additional oncogenic event[30]. Activation of the mechanistic target of rapamycin complex 1 (mTORC1) pathway as the preferred third driver event was observed in several in vivo studies[16,30] as well as in human ccRCC cases carrying VHL and PBRM1 mutations[30]. A key cellular complex, mTORC1 integrates nutrient and growth factor signaling to promote anabolic metabolism, supporting tumor growth and invasion[36]. REDD1, a transcriptional target of HIF1, suppresses mTORC1 activity via activation of TSC1/TSC2, functioning as a tumor suppressor checkpoint that limits the oncogenic potential of HIF1[37]. Thus, the activation of the mTORC1 pathway and abrogation of the intrinsic tumor suppressing activity of REDD1 and TSC1/2 may act as the final step to ccRCC development.
Ample clinical evidence support the importance of mTORC1 activation in the pathobiology of human ccRCC[38], and mTOR inhibitors provide known therapeutic benefit in the treatment of metastatic ccRCC[39]. Correspondingly, PBRM1 mutation has been associated with longer progression-free survival with everolimus, an mTOR inhibitor, vs. wild-type PBRM1 (12.8 months vs. 5.5 months)[40,41]. Similarly, patients carrying truncal mutations in the mTORC1 signaling pathway, including TSC1, TSC2, and mTOR, also benefited from mTOR inhibition[42,43]. Therefore, both clinical and preclinical data support the importance of loss of PBRM1 as the preferred second event and the activation of mTORC1 as the preferred second/third driver event in ccRCC tumorigenesis following VHL inactivation.
In particular to integrate mechanistic and therapeutic research completed on these pathways, we propose a model depicting the interconnection of VHL-HIF-PBRM1-TSC-mTORC1 in the development in ccRCC. This model involves three inactivating steps: (1) VHL loss; (2) VHL-PBRM1 loss; (3) VHL-PBRM1-TSC loss. The initial complete pathologic loss of VHL via chromosome 3p loss, mutations, and/or promoter methylation results in pseudohypoxia - a hypoxia-like molecular response despite normoxic conditions due to aberrant accumulation of HIF proteins. As observed in both human and mouse VHL loss models, the loss of VHL is insufficient in initiating ccRCC development[44]. The subsequent loss of PBRM1 signifies the second step HIF-PBRM1-STAT, which leads to a dysregulated feed-forward amplification loop of maximal downstream gene expression[30]. As evidenced by the PBRM1-/- VHL-/- mouse model, the activation of mTORC1 either by loss of TSC suppression or by aberrant increase in mTORC1 activity may encompass the third oncogenic step in ccRCC[30].
SETD2
SETD2 mutations are observed in 10% of human ccRCC primary tumors, and the frequency dramatically increase to ~30% in metastatic ccRCC patient samples[40], thereby representing an important molecular aberration in ccRCC metastatic progression[27,45]. SETD2 encodes a histone H3 lysine 36 methyltransferases that utilize a conserved SET domain[46]. Most significantly, SETD2-gene products are responsible for the trimethylation of the histone H3K36, loss of which is associated with widespread RNA processing defects, thereby affecting chromatin accessibility, transcriptional activation, DNA repair, and cell cycle regulation[47]. SETD2 functions as a tumor suppressor in several types of cancer, including breast cancer[48] and leukemia[49], and post-transcriptional regulation of SETD2 may be correlated with tumor development in breast cancer and ccRCC[50]; additionally, several studies suggest inactivating mutations of SETD2 occur most prevalently in ccRCC and non-small cell lung cancer[26,51].
SETD2 mutations are clinically associated with worse kidney cancer specific survival[27,52] and ccRCC metastasis[40], though SETD2 loss is not correlated with poor targeted treatment outcomes[40,53]. Tumor associated SETD2 mutations also highlight the presence of significant intratumor heterogeneity in ccRCC[1,54]. Multiple SETD2 mutation variants have been identified among different tumor regions in individual patients, suggesting its role in the convergent evolution of ccRCC[6,39]. Given its frequency of inactivation in ccRCC and its critical role as a tumor suppressor, loss of SETD2 function represents an important genomic progression marker in ccRCC.
BAP1
BAP1, like SETD2 and PBRM1, is found on chromosome 3p21. BAP1 encodes for a deubiquitinating enzyme, shown to bind breast cancer type 1 susceptibility protein (BRCA1) and BRCA1-associated RING domain protein 1 (BARD1) and thereby function as a tumor suppressor by inhibiting the ability of BRCA1/BARD1 to mediate ubiquitination and autoubiquitination[55,56]. BAP1 also regulates transcription via histone modulation, thus playing a key role in cell cycle and growth, cellular response to DNA damage, and chromatin dynamics[57]. BAP1 mutations are prevalent in about 10% of human ccRCC cases, and loss of BAP1 function is associated with tumors of high grade and large size as well as poor overall clinical outcome despite targeted therapy[27,28,40]. Kidney-specific homozygous deletion of VHL and BAP1 in mice resulted in early death, though some mice with heterozygous deletion of BAP1 and complete loss of VHL developed tumors only moderately resembling human ccRCC[58]. Further preclinical investigation of the BAP1 interaction with VHL inactivation is required to more clearly define the role of BAP1 in ccRCC tumorigenesis.
Current and emerging therapeutics
The management of metastatic ccRCC has improved dramatically over the last decade [Figure 2]. Prior to 2005, only two drugs were approved for the medical treatment of advanced ccRCC and the median survival was poor (15 months)[1]. The introduction of targeted therapies expanded the treatment options greatly and doubled the median survival (~30 months). Given the highly vascular nature of ccRCC, tyrosine kinase inhibitors targeting the VEGF signaling pathway provided considerable benefit over the interleukin-2 (IL-2) and interferon treatments. Seven anti-angiogenic drugs have been approved for first-line and second-line treatment of metastatic RCC since 2005, including sorafenib, sunitinib, pazopanib, axitinib, bevacizumab, cabozantinib, and lenvatinib[59-64]. With the exception of lenvatinib in combination with everolimus[61] and bevacizumab use with interferon-α[65], all approvals for VEGF-targeted therapies have been for single agents. The development of mTOR inhibitors, temsirolimus and everolimus, have provided additional therapeutic benefit as second-line single agents or in the first-line setting in patients with poor risk status[66,67]. Despite these therapeutic advances, the average duration of disease control with these drugs remains only 8-9 months for first line treatment and 5-6 months in the second line setting[1]. Additional target therapies under investigation also include PT2385, a first-in-class HIF-2α antagonist, which has shown a favorable safety profile and activity in a phase I dose escalation trial[9].
The new generation of immunotherapies has also shown promise for the treatment of metastatic ccRCC [Figure 2]. Immunotherapy has long been utilized to treat RCC with the use of interferon-α and high-dose IL-2[68], but these cytokine therapies were associated with significant toxicity and low response rates. Thus, the development of T-cell immune checkpoint inhibitors may similarly exploit intrinsic antitumor immune activity with fewer adverse effects and improved quality of life. Nivolumab, a monoclonal antibody against programmed cell death protein 1, was approved following the CheckMate 025 randomized control trial, which demonstrated an overall survival benefit of nivolumab over everolimus in patients who failed prior sunitinib or pazopanib therapy[69]. The results of CheckMate 214, a randomized clinical trial investigating nivolumab in combination with ipilimumab, an inhibitor of checkpoint cytotoxic T lymphocyte-associated protein 4 (CTLA4), vs. sunitinib in patients with previously untreated advanced ccRCC, were recently reported[70]. Overall survival and objective response rates of nivolumab plus ipilimumab were significantly higher than sunitinib among intermediate- and poor-risk patients. Thus, this combination has been granted by the FDA as a first-line therapy. Phase III clinical trials of additional combinations of checkpoint inhibitors with VEGF-targeted therapies are under investigation, which probably will again revolutionize the way we treat metastatic kidney cancer[1].
Conclusion
The fields of kidney cancer biology and therapy have undergone transformative changes over the past two decades. However, we are just at the beginning of contemplating how to best integrate these two seems distinct yet highly inter-informative disciplines. The exceptional clinical benefits derived from modern cancer therapy including targeted and immunotherapies have been associated with an astronomical socioeconomic burden to the society. In the authors’ opinion, the principal way for the medical society to further improve treatment efficacy and reduce the cost is the thoroughly integrate multi-omics based predictive biomarkers into standard practice for the selection of the most effective front-line/second line therapeutics with a curative intent. It is a tall but achievable order thus challenge awaiting to be conquered, exemplified are the shown transition of therapeutic eras in metastatic kidney cancer from the “Dark Age” (~2005, 2 drugs, median survival at ~15 moths) through the “Modern Age” (~2015, 9 drugs, median survival at ~30 months) to the “Golden Age” (2015~, 13 drugs and counting, 30 months and counting with significant number of patients expected to be cured). Nevertheless, much needs to be done if we wish to reach Diamond Age by 2025 when most kidney cancer patients could be rendered free of recurrence, progression, and thereby mortality from RCC[1].
Declarations
Authors’ contributionsConception, design of the study, and interpretation: Le VH, Hsieh JJ
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestDr. James J. Hsieh is a consultant for Eisai Inc.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, et al. Renal cell carcinoma. Nat Rev Dis Primers 2017;3:17009.
3. Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 2016;29:104-16.
4. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013;499:43-9.
5. Hsieh JJ, Le V, Cao D, Cheng E. Genomic classifications of renal cell carcinoma: a critical step towards the future personalized kidney cancer care with pan-omics precision. J Pathol 2018;244:525-37.
6. Hsieh JJ, Manley BJ, Khan N, Gao J, Carlo MI, et al. Overcome tumor heterogeneity-imposed therapeutic barriers through convergent genomic biomarker discovery: a braided cancer river model of kidney cancer. Semin Cell Dev Biol 2017;64:98-106.
7. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 2013;123:3664-71.
8. Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab 2014;2:3.
9. Courtney KD, Infante JR, Lam ET, Choueiri TK. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Onc 2018;36:867-74.
10. Hakimi AA, Tickoo SK, Jacobsen A, Sarungbam J, Sfakianos JP, et al. TCEB1-mutated renal cell carcinoma: a distinct genomic and morphological subtype. Mod Pathol 2015;28:845-53.
11. Kibel A, Iliopoulous O, DeCaprio JA, Kaelin WG Jr. Binding of the von Hippel-Lindau tumor suppressor protein to Elong B and C. Science 1995;269:1444-6.
12. Rohan SM, Xiao Y, Liang Y, Dudas ME, Al-Ahmadie HA, et al. Clear-cell papillary renal cell carcinoma: molecular and immunohistochemical analysis with emphasis on the von Hippel-Lindau gene and hypoxia-inducible factor pathway related proteins. Mod Pathol 2011;24:1207-20.
13. Carroll VA, Ashcroft M. Role of hypoxia-induced factor (HIF)-1 alpha versus HIF-2 alpha in regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the pathway. Cancer Res 2006;66:6264-70.
14. Shen C, Kaelin WG. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol 2013;23:18-25.
15. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010;29:625-34.
16. Fu L, Wang G, Shevchuk MM, Nanus DM, Gudas LJ. Generation of a mouse model of Von Hippel-Lindau kidney disease leading to renal cancers by expression of a constitutively active mutant of HIF1alpha. Cancer Res 2011;71:6848-56.
17. Fu L, Wang G, Shevchuk MM, Nanus DM, Gudas LJ. Activation of HIF2alpha in kidney proximal tubule cells causes abnormal glyogen deposition but not tumorgenesis. Cancer Res 2013;73:2916-25.
18. Kondo K, Clco J, Nakamura E, Kaelin WG. Inhibition of HIF is necessary for tumor suppression by von Hippel-Lindau protein. Cancer Cell 2002;1:237-46.
19. Zimmer M, Doucette D, Siddiqui N, Iliopoulous O. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL -/- tumors. Mol Cancer Res 2004;2:89-95.
20. Purdue MP, Johansson M, Zelenika D, Toro JR. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet 2011;43:60-5.
21. Kroeger N, Klatte T, Chamie K, Rao PN. Deletions of chromosomes 3p and 14q molecularly subclassify clear cell renal cell carcinoma. Cancer 2013;119:1547-54.
22. Motzer RJ, Jonasch E, Agarwal N, Bhayani S, Bro WP, et al. Kidney Cancer, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:804-34.
23. Rodgers JL, Bayeh L, Scheuermann TH, Longgood J, Key J, et al. Development of inhibitors of the PAS-B domain of the HIF-2α transcription factor. J Med Chem 2013;56:1739-47.
24. Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, et al. A small molecule antagonist of HIF-2α is efficacious in pre-clinical models of renal cell carcinoma. Cancer Res 2016;76:5491-500.
25. Kapitsinou PP, Haase VH. The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death Differ 2008;15:650-9.
26. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543-50.
27. Hakimi AA, Chen YB, Wren J, Gonen M, Abdel-Wahab O, et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinom. Eur Urol 2013;63:848-54.
28. Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavía-Jiménez A, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 2012;44:751-9.
29. Varela I, Tarpey P, Raine K, Huang D, Ong CK, et al. Exome sequencing identifies frequent mutation of SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011;469:539-42.
30. Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, et al. The SWI/SNF protein PBRM1 restrains VHL-loss-driven clear cell renal cell carcinoma. Cell Rep 2017;18:2893-906.
31. Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet 2014;166:350-66.
32. Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling genetics, genomics, and mechanisms. Cell Res 2011;21:396-420.
33. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013;45:592-601.
35. Gao W, Li W, Xiao T, Liu XS, Kaelin WG Jr. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL -/- clear cell renal carcinoma. Proc Natl Acad Sci USA 2017;114:1027-32.
36. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell 2017;169:361-71.
37. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/SC2 tumor suppressor complex. Genes Dev 2004;18:2893-904.
38. Robb V, Karbowniczek M, Szanto AJ, Henske EP. Activation of the mTOR signaling pathway in renal clear cell carcinoma. J Urol 2007;177:346-52.
39. Wei EY, Hsieh JJ. A river model to map convergent cancer evolution and guide therapy in RCC. Nat Rev Urol 2015;12:706-12.
40. Hsieh JJ, Chen D, Wang PI, Marker M, Redzematovic A, et al. Genomic biomarkers of a randomized trial comparing first-line everolimus and sunitinib in patients with metastatic renal cell carcinoma. Eur Urol 2017;71:405-14.
41. Hsieh JJ, Chen DPW, Chen YB, Redzematovic A, Marker M. Identification of efficacy biomarkers in a large metastatic renal cell carcinoma (mRCC) cohort through next generation sequencing (NGS): results from RECORD-3. J Clin Oncol 2015;33:abstr4509.
42. Voss MH, Hsieh JJ. Therapeutic guide for mTOuRing through the braided kidney cancer genomic river. Clin Cancer Res 2016;22:2320-2.
43. Kwiatkowski DJ, Choueiri TK, Fay AP, Rini BI, Thorner AR, et al. Muttions in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res 2016;22:2445-52.
44. Nargund AM, Ozmanbeyoglu HU, Cheng EH, Hsieh JJ. SWI/SNF tumor suppressor gene PBRM1/BAF180 in human clear cell kidney cancer. Mol Cell Oncol 2017;4:e1342747.
45. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying enzymes. Nature 2010;463:360-3.
46. Sun XJ, Wu J, Wei XY, Hu M, Wang L. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem 2005;280:35261-71.
47. Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res 2014;24:241-50.
48. Sarakbi AW, Sasi W, Jiang WG, Roberts T, Newbold RF, et al. The mRNA expression of SETD2 in human breast cancer: correlation with clinico-pathological parameters. BMC Cancer 2009;9:290.
49. Zhang J, ding L, Holmfeldt L, Wu G, Heatley SL, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukemia. Nature 2012;481:157-63.
50. Xiang W, He J, Huang C, Chen L, Tao D, et al. miR-106b-5p targets tumor suppressor gene SETD2 to inactive its function in clear cell renal cell carcinoma. Oncotarget 2015;6:4066-79.
51. Duns G, van-der-Berg E, van-Duivenbode I, Osinga J, Hollema H, et al. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal carcinoma. Cancer Res 2010;70:4287-91.
52. Manley BJ, Zabor EC, Casuscelli J, Tennenbaum DM, Redzematovic A, et al. Integration of recurrent somatic mutations with clinical outcomes: a pooled analysis of 1049 patients with clear cell renal cell carcinoma. Eur Urol Focus 2017;3:421-7.
53. Tennenbaum DM, Manley BJ, Zabor E, Becerra MF, Carlo MI, et al. Genomic alterations as predictors of survival among patients within a combined cohort with clear cell renal cell carcinoma undergoing cytoreductive nephrectomy. Urol Oncol 2017;35:532.
54. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883-92.
55. Nishikawa H, Wu W, Koike A, Kojima R, Gomi H, et al. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res 2009;69:111-9.
56. Mallery DL, Vandenberg CJ, Hiom K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J 2002;21:6755-62.
57. Piva F, Santoni M, Matrana MR, Satti S, Giulietti M, et al. BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: molecular diagnostics and possible targets for personalized therapies. Expert Rev Mol Diagn 2015;15:1201-10.
58. Wang SS, Gu YF, Wolff N, Stefanius K, Christie A, et al. Bap1 is essential for kidney function and cooperates with VHL in renal tumorigenesis. Proc Natl Acad Sci USA 2014;111:16538-43.
59. Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814-23.
60. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, et al. Sorafenib in advanced clear cell renal cell carcinoma. N Engl J Med 2007;357:203.
61. Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol 2015;16:1473-82.
62. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, et al. Sunitinb versus interferon alfa in metastatic renal cell carcinoma. N Engl J Med 2007;356:115-24.
63. Motzer RJ, McCann L, Deen K. Pazopanib versus sunitinib in renal cancer. N Engl J Med 2013;369:1970.
64. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931-9.
65. Escudier B, PLuzanska A, Koralewski P, Ravaud A, Bracarda S, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 2007;370:2103-11.
66. Hudes G, Carducci M, Tomczak P. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271-81.
67. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449-56.
68. McDermott DF, Regan MM, Clark JI, Flaherty LE, Weiss GR, et al. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J Clin Oncol 2005;23:133-41.
69. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803-13.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Le VH, Hsieh JJ. Genomics and genetics of clear cell renal cell carcinoma: a mini-review. J Transl Genet Genom 2018;2:17. http://dx.doi.org/10.20517/jtgg.2018.28
AMA Style
Le VH, Hsieh JJ. Genomics and genetics of clear cell renal cell carcinoma: a mini-review. Journal of Translational Genetics and Genomics. 2018; 2: 17. http://dx.doi.org/10.20517/jtgg.2018.28
Chicago/Turabian Style
Le, Valerie H., James J. Hsieh. 2018. "Genomics and genetics of clear cell renal cell carcinoma: a mini-review" Journal of Translational Genetics and Genomics. 2: 17. http://dx.doi.org/10.20517/jtgg.2018.28
ACS Style
Le, VH.; Hsieh JJ. Genomics and genetics of clear cell renal cell carcinoma: a mini-review. J. Transl. Genet. Genom. 2018, 2, 17. http://dx.doi.org/10.20517/jtgg.2018.28
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 34 clicks
Cite This Article 34 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.