
New approaches and prospects of immunotherapy and gene therapy for prostate cancer
 0
0 ,
, Abstract
Prostate cancer stands as the most prevalent cancer globally, constituting 21% of all cancer diagnoses in male patients. Urgent optimization of prostate cancer care is essential, given that this disease claims 345,000 lives every year. These innovative approaches hold substantial promise for both researchers and patients, representing a beacon of hope in the inhibitory act against prostate cancer. Prostate cancer's gradual advancement deems it suitable for immune therapy, but trials in metastatic cases show limited effectiveness, likely due to compromised immunity. Hindered by defective cellular responses, an immune-suppressive microenvironment, emerging evidence and breakthroughs, such as CAR-T therapy, inspire cautious optimism for advanced prostate cancer immunotherapy. Tumors utilize tactics to escape immune recognition, promoting the proliferation of MDSCs, Treg cells, and TAMs. Immunotherapy targets prostate cancer by mostly expressed target proteins and overexpressed target proteins. Immune cells play a role in tumor development and metastasis in advanced prostate cancer. Modulating the tumor microenvironment presents therapeutic possibilities. Certain prostate cancer types exhibit potential responses to immune checkpoint inhibitors, yet obstacles remain, necessitating additional research for enhanced efficacy. Immunotherapy faces hurdles in prostate cancer - limited inflammation, scarce antigens, and a resistant microenvironment. Grasping resistance intricacies is pivotal. The identification of DNA's helical structure propelled global progress in disease treatment through gene therapy. Choosing gene therapy vectors is critical; viruses are potent but toxic, while nonviral options, though less toxic, encounter barriers affecting transfection. In the realm of prostate cancer treatment, immunotherapy and gene therapy are emerging as increasingly viable options.
Keywords
INTRODUCTION
Prostate cancer ranks as the second most common type of human cancer globally, and in the United States alone, there were 288,300 new active cases and 34,700 deaths recorded in 2023. Prostate cancer constituted around 21% of male cancer cases that year[1]. While surgery, chemotherapy, and/or radiotherapy remain primary treatments for many solid tumors, combining immunotherapy with other medications is enhancing patient survival rates. Research advancements in immunotherapy for various solid tumors, including prostate cancer, show promising results. Prostate cancer studies indicate the significant role of inflammation in its growth and increment, with molecular heterogeneity defining the disease stages[2-5]. Capturing and knowing how immunity reacts to immunomodulatory drugs in prostate cancer can aid in developing innovative combination therapies. Typically, localized cases are managed with procedures such as radical prostatectomy or radiotherapy, followed by ongoing monitoring through PSA tests. There is a well-documented overall survival benefit of adding ADT to radiotherapy in localized prostate cancer. To date, prospective randomized trials have not investigated the use of ADT alongside stereotactic ablative radiotherapy (SABR). As such, there is no consensus as to the use and timing of ADT with SABR to treat hormone-sensitive oligometastatic prostate cancer. There is robust evidence indicating that androgen receptors activate DNA repair pathways, which provides a rationale behind the use of ADT with SABR for hormone-sensitive prostate oligo-metastases. Chronic inflammation, often linked to prostatitis-induced cellular and genetic damage, strongly influences prostate cancer development and progression[6,7].
Prostate cancer is often termed a “cold tumor” due to its immunosuppressive surroundings. In this environment, infiltrating lymphocytes hinder the activeness of T-effector cells, promoting the increment of prostate cancer. Biopsy samples reveal that these lymphocytes typically exhibit T helper 17 and T regulatory phenotypes, which impede the patient’s immune response against tumor and the production of autoreactive T cells[8,9]. Prostate cancer with weaker T-cell shortlisting and filtration power, a less suppressive tumor microenvironment (TME), and fewer mutations is less responsive to immunotherapy. Despite these challenges, a subset of prostate cancer patients display immunogenic traits. Recent examples of positive responses to immunosuppressive drugs (ISDs) or their combinations include patients with more expression of PD-L1 tumor, CDK12 mutational changes, significant tumor-mutational burden, high microsatellite instability (MSI) cancers, mismatch repair-deficient (dMMR) individuals[10,11]. However, unlike head and neck cancer, non-small-cell lung cancer, melanoma, and renal cell carcinoma, prostate cancer shows limited success in immunity responses to treatment due to its immunosuppressive nature[12]. Biallelic inactivation of CDK12 is associated with a unique genome instability phenotype. The CDK12-specific focal tandem duplications can lead to the differential expression of oncogenic drivers, such as CCND1 and CDK4[13]. As such, there is a possibility of vulnerability to CDK4/6 inhibitors for CDK12-mutated tumors. Moreover, the CDK12 aberrations may be used next to mismatch repair deficiency, as a biomarker of treatment response[14]. This highlights the rationale for the combination therapeutic strategy of immune checkpoint blockade and CDK4/6 inhibition in clinical trials[15-17]. Immunotherapy trials aim to target T cell infiltration and the mutational load of prostate cancer cells, and harness the combined power of treatments to counteract the inhibitory tumor microenvironment (TME)[10,18].
Gene therapy involves the use of specialized medications to target specific genes, either by modifying the genetic code responsible for certain outcomes or by altering tissue characteristics, with the aim of treating various illnesses. Initially, gene therapy focused on simple genetic disorders such as severe combined immunodeficiency, aiming to replace defective genes[19,20]. So far, the advent of cancer gene therapy brought new perspectives and techniques, recognizing cancer as a condition involving both germ cell and somatic cell genetic changes[21]. Prostate cancer, particularly early-stage cases detectable through blood tests, can be targeted effectively using gene therapies, especially via intra-prostatic injections. This method allows precise anatomical targeting, benefiting both direct cytotoxic and immunotherapy-based gene therapies[22,23]. Moreover, certain solid tumors, including prostate cancer, exhibit overexpression of the osteocalcin gene, making it a significant target for gene therapy interventions[24].
The way a patient responds to treatment is affected by various factors, including intra-tumor differences and previous therapies. This highlights the need for personalized and combined treatments, emphasizing their vital role in future strategies for successful immunotherapy and gene therapy. This overview delves into current and emerging treatments for prostate cancer, with a focus on immunotherapy and gene therapy. It addresses challenges posed by the unique immunosuppressive tumor microenvironment, discussing active and passive immunotherapy, adoptive T-cell treatment, and immune checkpoint inhibitors. The review explores the roles of immune cells (MDSCs, Tregs, and TAMs) in prostate cancer progression and treatment resistance and identifies key target proteins and antigens. Additionally, it provides insights into gene therapy, encompassing gene editing techniques and delivery methods.
CURRENT CHALLENGES IN IMMUNOTHERAPY RESPONSES IN PROSTATE CANCER
Prostate cancer progresses slowly compared to other malignancies, rendering it an optimal target for immune therapy. However, clinical trials employing various immune therapy methods, such as active immunotherapy, passive immunotherapy, adoptive T-cell treatment, and the combination of immune checkpoint inhibitors with chemotherapy, have shown limited effectiveness in metastatic castration-resistant prostate cancer (mCRPC)[25]. The in-effectiveness of recent immune therapy in metastatic prostate cancer might stem from the compromised immune system in these patients[26]. They often exhibit defective cellular immunity, reduced natural killer (NK) cell activity, and lower circulating T-cell frequencies[27]. The tumor microenvironment in prostate lesions creates an unfavorable niche for immune cells[28-30], limiting the efficacy of immunotherapy[31,32]. Studies have indicated reduced infiltration of tumor-infiltrating CD8+ T cells in patients treated with antiandrogen like abiraterone[33]. Immune checkpoint inhibitors, although they block PD-1 and PD-L1 interactions, face challenges due to different kinds of immune-suppressive traits within the prostate tumor microenvironment, such as higher plasma TGF-β concentration and increased suppressive cells like TAM, Tregs, and MDSCs[34-38]. Prostate cancer often exhibits limited infiltration of efficient immune cells, referred to as a “cold” tumor, due to weakened cellular immunity and a highly immune-suppressive tumor microenvironment. It is unclear whether the absence of immune infiltration stems from the failure of effector natural killer cells and T cells to home in on the tumor. Additional potential resistance pathways have been suggested, including immunological tolerance[39,40] and decreased mutational tumor load, indicating resistance to immunotherapy in male subjects with prostate cancer[41].
Several Phase-III clinical trials and active immunotherapy trials have been conducted for prostate cancer subjects, although their effectiveness remains limited. Emerging evidence from small-scale clinical trials has shown promise, and CAR-T therapy breakthroughs have transformed the treatment landscape for refractory malignancies. Prostate cancer's pleiotropic effects, including leukocyte infiltration, hormonal escape, angiogenesis, development, and endothelial-mesenchymal transition, are linked to cytokines and chemokines. Targeting the chemokine system and immune cells is essential to developing effective immunotherapies for prostate cancer. Despite challenges, there is cautious optimism about the future of immunotherapy for advanced prostate cancer[42,43].
IMMUNE EVASION IN PROSTATE CANCER
Tumors have evolved ways to prevent identification by the immune system. Myeloid-derived suppressor cells (MDSCs), T regulatory cells (Tregs), and tumor-associated macrophages (TAMs), which block effector T-cell functions, can all be attracted to and grow in the tumor microenvironment[44,45] [Figure 1].

Figure 1. Immune Evasion in Prostate Cancer, the inhibitory effect of MDSCs, Tregs, and TAMs on effector T-cell functions. The activated T Cells (aATCs) with bispecific antibodies target MDSCs and inhibit their suppressive function and show the inhibition of MDSC-associated enzymes and the release of cytokines and chemokines. Therapeutic approaches such as vaccines and therapeutic agents (imatinib, sunitinib, cyclophosphamide, gemcitabine) target the immunosuppressive microenvironment. The Th17 cells producing IL-17 as pro-inflammatory cells and the frequency of CCR4/IL-17/CD4+ T cells in prostate cancer patients increases the immunotherapy and antitumor response. Negative costimulatory ligands (PDL-1, CTLA-4), regulatory lymphocytes, myeloid cells, and immunosuppressive substances (IL-10, TGF-β, IDO) show inhibitory effects on immune cells.
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells are the major subset of cells that play a role in the immunosuppressive tumor microenvironment[46,47]. There are various factors that contribute to MDSC accumulation and activation, many of which have been linked to chronic inflammation[48,49]. The growth of MDSCs is regulated by different kinds of inflammatory mediators, and STAT3 is probably the most important transcription factor in this process[50]. A significant percentage of CD14+/HLA-DRlow/- monocytic MDSCs was found in treated PCa patients (30.7 15.0% CD14+ cells) compared to untreated PCa patients (10.6 14.3%, P = 0.0001) in an analysis of changes in the levels of circulating MDSCs with PCa progression, following immune-therapy. In vitro, these CD14+/HLA-DRlow/- monocytes were effective at inhibiting immune cell activity. Thus, eliminating these MDSCs may dramatically enhance the benefits of cancer immunotherapy and antitumor responses[51,52]. In a phase II trial, researchers investigated the efficacy of the anticancer drug tasquinimod (TASQ) in males with metastatic castration-resistant prostate cancer (CRPC) with limited symptoms. Myeloid-derived suppressor cells (MDSCs), which encourage tumor growth and dissemination, are the target of TASQ because they express the S100A9 receptor. During the trial, patients were randomly assigned in a 2:1 ratio to receive either TASQ or a placebo. TASQ was administered orally once daily, commencing at a dose of 0.25 mg/d and gradually increasing to 1.0 mg/d over the course of 4 weeks. The primary outcome was the percentage of patients who showed no disease progression at six months. 201 patients with similar baseline characteristics were enrolled in the trial, of whom 134 received TASQ, while 67 received a placebo. According to the findings, TASQ outperformed placebo in terms of both the median progression-free survival (PFS) (7.6 vs. 3.3 months, P = 0.0042) and the 6-month progression-free proportion (69% vs. 37%, P = 0.001). The trial found that TASQ had a tolerable side effect profile, reduced disease progression in patients with metastatic CRPC, and improved PFS. The trial also indicated that TASQ exerted antiangiogenic and antimetastatic effects by altering MDSC activity in the tumor microenvironment[53-56]. Utilizing activated T cells (aATCs) that are equipped with bispecific antibodies (Bi) against tumor antigens like Her2 or EGFR has been demonstrated to increase the antitumor effects of immunotherapy. Additionally, to directly destroy tumor cells, these aATCs can lower the quantity and activity of myeloid-derived suppressor cells (MDSCs), which are immune cells that block the antitumor immune response. The expression of enzymes like COX2, PGE2, and ARG1 that mediate the suppressive function of MDSCs can be inhibited by aATCs. Additionally, aATCs have the ability to create cytokines and chemokines such as IL-2, IFN-, CXCL9, and CXCL10 that aid in the attraction and activation of other immune cells. Consequently, this approach can concurrently target tumor cells and MDSCs, which will improve the final outcome of immunotherapy[57].
Tumor microenvironment modulation to enhance immune-based therapies
Tumors can employ a variety of tactics to avoid immune attack and establish a tolerant microenvironment. These tactics include reducing antigen presentation, activating unfavorable costimulatory signals, creating immunosuppressive substances, enlisting regulatory cells, etc. These mechanisms can inhibit the activity and function of different types of immune cells, including dendritic cells, natural killer cells, and T cells. The presence of negative costimulatory ligands such as PDL-1 and CTLA-4, along with regulatory lymphocytes and myeloid cells, as well as tumor-derived factors such as IL-10, transforming growth factor-β (TGF-β), and IDO, presents challenges for effectiveness of immune-therapy and antitumor actions[58,59]. To overcome these challenges, combining vaccines with therapeutic approaches designed to counteract the immune-suppressive microenvironment, like using imatinib (to inhibit IDO), sunitinib (to counteract MDSCs and Treg cells), cyclophosphamide (to eliminate Treg cells), gemcitabine (to eliminate MDSCs), can increase the impact of immunotherapy, bolster antitumor immune responses[60-63].
T-regulatory and T-17 cells
Tregs are immune cells that suppress the immune response to self-antigens and tumors, while Th17 cells are immune cells that produce a pro-inflammatory cytokine called IL-17. Peripheral tolerance to self-antigens is regulated by Tregs, constituting 5%-10% peripheral CD4+ T cells. Treg deficiency can lead to autoimmune responses, and these cells play a crucial role in dampening the immune system's response to cancers, thereby facilitating tumor growth. Enhanced immune suppression in prostate cancer patients is linked to tumor development. Following androgen ablation, an increase in Tregs might contribute to the temporary immune response. Studies comparing pre- and post-vaccination patients revealed a correlation of P = 0.029, within overall survival (OS), and a decrease in Treg suppressive activity[64-68]. Prostate cancer patients undergoing active whole-cell immunotherapy displayed an inverse relationship between progression-free survival (TTP) and the frequency of CCR4/IL-17/CD4+ T cells before immunization. Responders had Th17 profiles similar to healthy controls, differing significantly from non-responders. In mice with endogenous prostate cancers, adding less dose of cyclophosphamide to cell-based immunotherapy enhanced treatment effectiveness by modulating Teff/Treg ratios, suppressing Tregs and boosting effector T cells. FLII, controlling PD-L1 expression via the YBX1 signaling axis, is vital in enzalutamide-resistant CRPC. Inhibiting this pathway synergistically enhanced CRPC treatment, reducing Tregs and MDSCs while promoting CD8 T cell proliferation. These findings support targeted therapy for endocrine therapy-resistant CRPC, utilizing the functional link between signaling pathways of FLII, YBX1/PD-L1[69-71].
TARGETS FOR PROSTATE CANCER IMMUNOTHERAPY
Various forms of immunotherapy are available for treating prostate cancer. The following are some immunotherapy targets for prostate cancer [Figure 2].
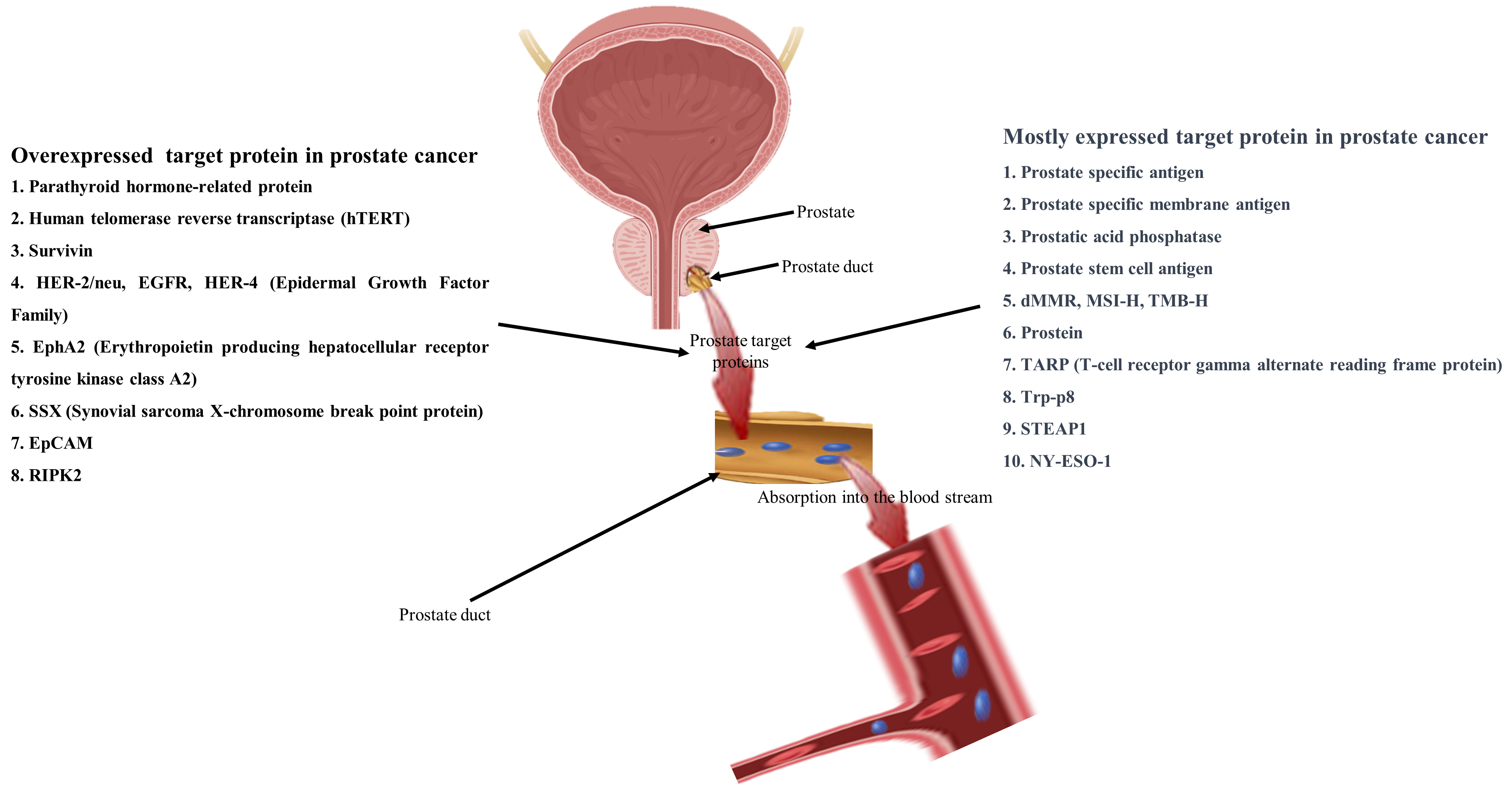
Figure 2. Schematic view of target proteins for immunotherapy, mostly expressed and over-expressed in prostate cancer. Prostate cancer mostly expressed target proteins for immunotherapy such as prostate-specific antigen (PSA), PSMA (prostate-specific membrane antigen), prostatic acid phosphatase, PSCA (Prostate stem cell antigen), dMMR (DNA mismatch repair deficiency), MSI-H (microsatellite instability), TMB-H (high tumor mutational burden), prostein, TARP (T-cell receptor gamma alternate reading frame protein), Transient receptor potential melastatin 8 (Trp-p8), six transmembrane epithelial antigen of prostate-1 (STEAP1), NY-ESO-1 and overexpressed proteins such as parathyroid hormone-related protein, human telomerase reverse transcriptase (hTERT), survivin, HER-2/neu, EGFR, HER-4 (Epidermal Growth Factor Family), EphA2 (Erythropoietin producing hepatocellular receptor tyrosine kinase class A2), SSX (Synovial sarcoma X-chromosome break point protein), EpCAM(epithelial cell adhesion molecule), RIPK2 (receptor-interacting protein kinase 2).
Mostly expressed target proteins in prostate tissues
Prostate-specific antigen (PSA), a protein exclusive to prostate; prostate-specific membrane antigen (PSMA), a membrane protein present in prostate; prostatic acid phosphatase (PAP), an enzyme associated with prostate; prostate stem cell antigen (PSCA), an antigen linked to prostate stem cells; prostein, a protein specific to prostate; TARP (T cell-receptor gamma alternate reading frame protein), a peptide involved in T cell receptor signaling; Trp-p8, a protein relevant to prostate cancer; six-transmembrane epithelial antigen of the prostate 1 (STEAP1), a transmembrane protein associated with prostate; NY-ESO-1, a specific antigen with relevance to tumors [Figure 2].
Prostate-specific antigen
The presence of prostate-specific antigen (PSA) in a high proportion of prostate cancer (PCa) tissues makes it a commonly applicable serum marker for identifying and monitoring PCa. PSA, a serin-protease similar to kallikrein, is usually displayed in prostate epithelial cells[72-74]. Studies have identified PSA-derived peptides that activate tumor-reactive CD8+ cytotoxic T lymphocytes (CTLs) when presented by human leukocyte antigen-A2 (HLA-A2) and HLA-A3[75-77]. Simultaneous induction of tumor-reactive CTLs and HLA-A2/A3-restricted epitopes of PSA has been achieved using specific oligopeptides[78-80]. Additionally, HLA-A24-restricted PSA peptides elicited peptide-specific CTLs in PCa patients and HLA-A*2402-restricted CTLs in transgenic mice[81-83]. The role of various HLA class I and II molecules is to present immunogenic PSA peptides[84,85]. Efforts to optimize active immunotherapy delivery methods have gained attention, such as using adeno-associated virus-based vectors to transduce dendritic cells (DCs). These modified DCs stimulated PSA-specific CTLs more efficiently than protein-pulsed DCs In vitro[86]. In mouse models, PSA has successfully triggered specific T cell responses. Researchers have explored binding vaccination techniques by androgen deprivation, showing enhanced CTL responses in HLA-A*0201/human PSA-twice transgenic mice when castrated before immunization by a PSA-presenting vaccinia virus. Androgen ablation was found to reduce CD4+T cell tolerance to prostate-specific antigens, suggesting that targeted immunotherapy for PCa might be more effective post androgen ablation[87,88].
Typically, patients with increased PSA levels are transferred to a urologist for identifying and clarifying testing, which could be a prostate MRI or a biopsy[89]. Among the ten studies that were included, a variety of estimates for PSA's accuracy were discovered. However, the methodologies of the included studies were only vaguely described in that review, and it was significantly unclear whether any of them assessed PSA in patients who were symptomatic or asymptomatic or whether any of them were applied to primary care populations. In a review of the literature, researchers noted the dearth of studies addressing the first level of care and the majority of prostate-specific antigen tests are conducted[90]
Prostate-specific membrane antigen
Prostate-specific membrane antigen (PSMA) was initially discovered in various natural active tissues such as the brain, salivary gland, breast epithelium, renal tubular epithelium, and small intestine. While it was highly tissue-specific, non-prostatic tissues had significantly lower expression levels, ranging from 100 to 1,000 times lower. In prostate cancers, especially in advanced undifferentiated metastatic hormone-refractory prostate cancer (HRPC), PSMA serves as a marker/identifier for healthy prostate cells and is commonly present in major prostate tumors. Studies, both in vivo and In vitro, have explored the optimal antigen to trigger T cell responses, demonstrating that co-transducing genes encoding the extracellular portion of PSMA, a costimulatory peptide, with an adenoviral vector, efficiently triggers targeted T cell responses and fosters immune reactions against tumors in murine models[91-95]. PSMA has also emerged as a promising target for antibody treatment due to its surface expression on prostate cancer cells. Various anti-PSMA monoclonal antibodies, such as J591, linked to ricin A, bismuth-conjugated mab J591, have exhibited target-specific cytotoxicity against PSMA-expressing prostate cancer cells. Additionally, radioimmunotherapy employing antibody-conjugates of J415 and J591, bound to radioactive isotopes, has demonstrated preferential accumulation in live tumor regions in xenograft models, leading to potent and targeted anticancer effects both in vitro and in vivo[96-101]. Furthermore, PSMA has become a recognized target for prostate cancer treatment, with recent FDA approval of 177Lu-PSMA-617 for the treatment of advanced metastatic castrate-resistant prostate cancer (mCRPC) following successful outcomes in phase III VISION trials. There are ongoing developments in PSMA-targeted radiopharmaceuticals, including substances like J591 and TLX591 that utilize monoclonal antibodies to target PSMA. Additionally, small compounds such as PSMA 617, PSMA T&I, and MIP 1095 are being employed to target PSMA in these contexts[102].
Prostatic acid phosphatase
The principal protein released by prostate epithelial cells is PAP (Prostatic acid phosphatase), primarily found in benign and cancerous prostate tissue. Related identifications indicate low PAP mRNA display in non-prostate specific tissues such as kidney, testis, and placenta[66]. Immunogenic PAP-derived peptides binding with HLA-A2 were discovered, leading to specific tumor rejection in vivo[103-106]. Vaccination techniques using PAP target antigen showed immunotherapeutic potential, activating PAP-CD8+ cytotoxic T-lymphocytes and inhibiting tumor growth in animal models[107-110]. Sipuleucel-T, a binding together protein of GM-CSF and PAP, demonstrated effectiveness in phase III trials for advanced prostate cancer patients. Patients receiving sipuleucel-T had a 22% lower risk of dying and a median survival of 25.8 months compared with 21.7 months in placebo group. Researchers explored whether subsequent doses of a DNA vaccine coding for PAP could augment PAP-specific T-cell activity after sipuleucel-T treatment[110,111]. Despite an equal half increase in survival of four months, this is a competitor for drugs such as docetaxel, abiraterone, radium 223, cabazitaxel, and enzalutamide, which are licensed for this stage of prostate cancer[112-114]. A better outcome in the longer lifespan may have been achieved in subjects with less disease pressure and proof of immune response to the PAP (prostatic acid phosphatase) antigen potent in relation to anyone like antigen-specific IgG and T cells[115-117]. The study revealed a stronger Th2 response with DNA immunization, suggesting potential benefits from administering DNA immunization before sipuleucel-T[113,119,120]. Despite advancements, surgery remains crucial for PAP-positive prostate cancer patients, indicating the importance of monitoring serum PAP levels in clinical practice[117,120].
Prostate stem cell antigen
In research and studies conducted by researchers, HLA-A2-restricted PSCA peptides have been identified to induce In vitro tumor-reactive CTL responses[121-123]. Elevated levels of CD8+ T cell lymphocytes recognize these peptides which were found in the blood of prostate cancer subjects. Additionally, an HLA-A24-presented peptide stimulating CTLs in PCa subjects was discovered[124]. The TRAMP mouse model, which mimics the production and display of PSCA during prostate cancer progression, was used to study the immunotherapeutic potential of PSCA. Vaccination with a viral vector expressing PSCA, following priming by PSCA cDNA, significantly increased survival rates in TRAMP mice in the presence of prostate intraepithelial neoplasia compared to the control group[125].
Moreover, in another study utilizing this mouse model, recombinant DNA with modified vaccinia virus Ankara vectors, which codes for PSCA, STEAP1, prevented PCa progression[126]. Treatment with unbound anti-PSCA antibody 1G8 reduced metastasis formation, increased long-term survival, and slowed xenograft growth. Mechanistic studies revealed that target cross-linking was necessary for the Fc-independent induction of cell destruction and death. PSCA is considered a target for immunotherapy using antibodies. Mice with xenografts experienced cytotoxicity and remission after receiving anti-PSCA mAbs combined with the toxin maytansinoid[127-129].
Chimeric and human anti-PSCA antibody radio conjugates specifically target xenografts that are PSCA-positive and display anticancer effects in vivo. Moreover, bispecific antibodies designed to target prostate stem cell antigen and CD3 on human T cells activate immune-effector cells, leading to the elimination of tumor cells[129-131]. Altered T cells expressing chimeric-antigen receptors targeting PSCA have demonstrated high efficacy in lysing PSCA-expressing cells. In a recent study using an immunocompetent mice model of prostate and pancreatic tumors expressing PSCA, the safety and effectiveness of PSCA-CAR T cells were evaluated. Researchers observed both safety and long-lasting antitumor immune responses with PSCA-CAR T cells, despite PSCA expression in various organs, such as stomach, prostate, and bladder. All preclinical experiments highlight the potential of harnessing endogenous immunity against PSCA+ prostate cancers. Ongoing clinical trials are evaluating PSCA-targeted CAR-T cell therapies, along with a study involving CAR-T cells and BiTE antibodies[132-134].
DNA mismatch repair deficiency, microsatellite instability, high tumor mutational burden
DNA mismatch repair deficiency (dMMR), microsatellite instability (MSI-H), and increased tumor load of mutations are significant for cancer biology and therapy, because they influence the production and identification of neoantigens, which are new peptides formed from mutant proteins presented to the immune system by tumor cells. Neoantigens can trigger an antitumor immune response with stimulation of T cells, a subset of immune cells capable of removing cancer cells. However, tumor cells sometimes employ various strategies to evade or suppress the immune system. The detection of MSI-related prostate cancer can be improved for better quality or accuracy through IHC to find mismatch repair-related proteins, microsatellite instability analysis, and next-generation sequencing. Despite the rarity of this kind of event, it has therapeutic value. Additionally, a study found that bladder cancer subjects with DNA mismatch repair deficiency/microsatellite instability genotypes have a favorable prognosis for anti-PD-1/PD-L1 therapy[135-138].
Prostein
Prostein is a transmembrane protein that is commonly produced in both healthy and cancerous prostate tissues and may play a role in prostate cancer cell invasion and migration. Our research indicates that 87% of the primary tumors have maintained or even raised transcript levels compared to autologous non-malignant tissue samples. When compared to cancers that are not organ-confined, prostein expression is notably higher in prostate cancer[138-142]. Researchers have discovered a peptide derived from prostein and presented by HLA-A*0201, which activates tumor-reactive cytotoxic T lymphocytes (CTLs) when CD8+ T cells are stimulated In vitro with dendritic cells filled with the peptide, and also found immunogenic T-cell epitopes, such as HLA-Cw*0501 and HLA-B*5101[111,140,143].
According to findings by Wolfgang et al., prostein is not detectable in non-prostatic glands but can be identified using immunohistochemical (IHC) methods in acinar, intra-ductal prostate-gland adenocarcinomas[144].
T-cell receptor gamma alternate reading frame protein
The non-rearranged T-cell receptor gamma-chain locus has a unique androgen-regulated gene that gives rise to T-cell receptor gamma alternate reading frame protein (TARP). Mitochondria of PCa in men exhibit TARP. Females with breast cancer can be identified by TARP[145,146]. Researchers discovered that androgen upregulated TARP in the mitochondria of prostate cell lines using subcellular fractionation, immunocytochemistry, and Western blot analysis. Immunohistochemistry and mitochondrial fractionation data suggested that TARP was present in the outer mitochondrial membrane and that it might interact with mitochondria to carry out its biological activity[147].
In vitro prostate and breast cancer cell-reactive cytotoxic T lymphocytes are activated by a number of naturally occurring HLA-A*0201-restricted TARP peptides. Additionally, 2HLA class-II binding amino acid residues generated with TARP have been demonstrated to trigger efficient CD4+ T cell responses[148-150]. In order to evaluate TARP for both therapeutic and diagnostic purposes, a new targeted strategy using antibodies that bind each HLA class-I peptide group on tumor cell surfaces.
The possibility of incorrect matching between the exogenous, endogenous T-cell receptor and T-cell receptor chains is a significant drawback of employing TCR-engineered T cells. TCRs reactive against self-antigens and TCRs with unexpected specificity could result from mispairing, which would produce autoreactive T cells. Additionally, incorrectly paired transferred TCRs may compete with one another for CD3, lowering the level of surface expression. There are a number of methods to prevent mispairing, such as murinizing human TCRs to give them a comparative benefit for the interaction of CD3[151-155].
Transient receptor potential melastatin 8
Transient receptor potential melastatin 8 (TRPM8) is a calcium channel amino acid that is encoded by gene TRPM8 and activated by cold temperatures, menthol, and other substances[156]. TRPM8 is primarily expressed in sensory neurons, which are responsible for mediating the cold sensation[157]. Additionally, TRPM8 is also expressed in non-neuronal organs, such as the prostate gland[158]. Androgens, the male sex hormones that increase the growth and functionality of the prostate, control TRPM8 expression inside the prostate[158]. TRPM8 is expressed more in stromal cells compared to normal epithelial prostate cells, particularly androgen-sensitive cells. Although the activity of TRPM8 in prostate cancer is not clear, some studies imply that it may have functional activity in cell survival, invasion, and metastasis[158-160]. Consequently, it is a therapeutic target for the cure of prostate cancer and a possible marker for disease identification[157-160]. A seven-span transmembrane protein, exhibiting a significant resemblance to Ca2+ channel proteins, is encoded by the TRPM8 gene. Most prostate cancers exhibit the expression of TRPM8, primarily limited in the prostate[156]. Upon investigating differences from equivalent normal prostate tissue, upregulation of it was observed in early-stage cancers. We discovered an HLA-A*0201-binding protein that might incite tumor-reactive CTLs in vitro[111,161].
Six transmembrane epithelial antigen of prostate-1
Iron and copper metabolism, as well as other cellular functions, are affected by the protein known as Six transmembrane epithelial antigen of prostate-1 (STEAP1)[162]. The protein is typically overexpressed in prostate cancer cells, particularly those susceptible to androgens, the male sex hormones[163,164]. While STEAP1 is usually expressed at low levels in several organs, it plays a significant role in influencing epithelial-mesenchymal transition (EMT), leading to changes in the activities of cancer cells, making them more mobile and aggressive. Consequently, STEAP1 may contribute to the growth, invasion, and metastatic spread of prostate cancer[162,163]. Thus, it is plausible that STEAP1 could serve as both a biological marker and a therapeutic target for prostate cancer[162,163]. Moreover, researchers have discovered three promiscuous epitopes of CD4+ T cells and a various naturally occurring HLA-A2 restricted protein that can induce cytotoxic T-lymphocytes in vivo and in vitro[106,165-169]. Some immunization techniques, utilizing viral or recombinant cDNA vectors that code for mouse six-transmembrane epithelial antigen of prostate-1, have shown increased efficacy in inducing particular T cell expression, slowing tumor development, and lengthening longevity in mice models[126,170,171].
Recent research indicates that STEAP1 may also be a promising target for immunotherapy using antibodies; a pair of STEAP-specific monoclonal antibodies (mAbs) dramatically slowed the progression of PCa xenografts (mice)[172].
NY-ESO-1
Nine out of twenty-three (39%) prostate cancer patients had NY-ESO-1 mRNA expression. In 12 of 23 (52%) PCa patient sera and in 5 of 9 (55%) tumors expressing NY-ESO-1, antibodies against the protein were identified. However, neither mRNA copies nor NY-ESO-1 were identified in any of the studied BPH patients[173]. NY-ESO-1, a cancer testis antigen (CTA), is a peptide typically solely observed in the testis but can occasionally be found in other malignancies, such as prostate cancer. NY-ESO-1 is a significant immunogenic cancer testis antigen, which means that it can stimulate the body's production of antibodies and T cells[174]. Therefore, NY-ESO-1 might be a helpful immunotherapeutic target in cancer treatment that leverages the immune system[174]. Research revealed that 20 of 53 (38%) prostate cancer specimens had NY-ESO-1 mRNA expression, with a higher prevalence observed in more advanced disease stages. While none of the 78 subjects with localized prostate cancer had NY-ESO-1 antibodies, 10 of the 140 (7.1%) patients with metastatic prostate cancer did. Some patients who had antibodies against NY-ESO-1 also showed CD8 T cell responsiveness specific to NY-ESO-1. These results imply that prostate cancer progression and aggressiveness are related to NY-ESO-1 expression and immunogenicity[175].
For subjects with prostate cancer, numerous clinical trials have been conducted or are currently underway to evaluate the safety and efficacy of immunotherapy centered around NY-ESO-1. These trials encompass various approaches, including immunization with recombinant NY-ESO-1 peptide in combination with the adjuvant CpG 79093[176], which boosts immune responses, or using the viral vector MVA-NY-ESO-1[174], which transmits the desired gene. All these studies have shown that the NY-ESO-1 vaccine can stimulate or elicit NY-ESO-1-specific immune responses in prostate cancer subjects, which may have certain therapeutic advantages, such as tumor regression or disease stabilization[173,176]. However, to establish the long-term results and ideal practices of NY-ESO-1 immunotherapy for prostate cancer, more research is required.
Overexpressed proteins in prostate cancer, including tumors
Many proteins, including those associated with prostate cancer, exhibit overexpression across various cancers. Overexpression occurs when cells produce an excess amount of a specific protein beyond what is required. This phenomenon can significantly influence the growth, survival, and functionality of cells. Several of the proteins that are overexpressed in prostate cancer include [Figure 2]:
Parathyroid hormone-related protein
PTHrP plays a crucial role in bone development by binding to receptors on osteoblasts. Its involvement in the formation of bone metastases is particularly pronounced in PCa and other epithelial-origin malignancies[177,178]. Numerous prostate cancer cell lines and metastatic bone lesions exhibit the presence of PTHrP. Through activation of several pathways and interaction with other molecules, PTHrP can promote tumor growth, survival, invasion, and metastasis. Additionally, PTHrP can confer resistance to apoptosis in prostate cancer cells, which is triggered by various stimuli, including chemotherapy or radiation. By stimulating angiogenesis and osteolysis (breakdown of bones), PTHrP has an impact on the TME (tumor microenvironment)[179,180].
The role of PTHrP in prostate cancer is complex and not fully understood. However, it is obviously implicated in many facets of prostate cancer initiation and progression. As a result, PTHrP may serve as a potent biological marker for assessing the potential malignancy of prostate cancer, as well as a promising target for therapy aimed at retarding tumor growth and improving patient survival.
Human telomerase reverse transcriptase
In the majority of human malignancies, there is an overexpression of human telomerase reverse transcriptase (hTERT) is overexpressed, making it a crucial target for cancer therapeutics. Most non-transformed somatic cells lack this molecule, although it is present in over 85% of human malignancies, including PCa[181]. Research has identified a number of naturally occurring restricted epitopes to HLA-A*0201 of CTL that effectively generate peptide-specific and tumor-lysing CTLs in both in vitro and in vivo settings[111,182].
Additionally, in the initial instances of hTERT-based immunization experiments, direct evidence emerged linking the formation of tumor-infiltrating T cells to positive clinical outcomes. A number of clinical trials were initiated and are ongoing to evaluate the ability of hTERT to treat prostate cancer[183,184].
Dendritic cells (DCs), specialized antigen-presenting cells, are used in cell-based vaccination to introduce hTERT antigens to the immune system. To effectively induce anti-hTERT immunity, DCs can be loaded with hTERT peptides, mRNA, or DNA, and then administered to patients. This tactic has also been put to the test in prostate cancer clinical trials[183,184]. TERT inhibitors are tiny compounds that bind specifically to the hTERT catalytic site, thereby inhibiting its function, shortening telomeres and inducing apoptosis or cellular senescence. Examples of TERT inhibitors include substances like BIBR1532, MST-312, and GRN163L[185,186]. G-quadruplex stabilizers are substances that bind to the G-rich sequences in telomeric DNA, leading to the formation of G-quadruplexes, four-stranded structures that block hTERT from accessing the telomeres and suppress its activity. G-quadruplex stabilizers include BRACO-19, RHPS4, TMPyP4, etc.[185,187]. Epigenetic modulators are medications that change the methylation or acetylation of the hTERT gene promoter region to modify the chromatin structure and gene expression. Trichostatin A (TSA), 5-aza-2'-deoxycytidine (DAC), and valproic acid (VPA) are a few examples of epigenetic modulators[187]. Signaling system inhibitors are drugs that target upstream or downstream signaling pathways, such as PI3K/AKT/mTOR, catenin /- Wnt, NF-B, and MAPK, which regulate hTERT expression or activity[187].
Therefore, further investigation and improvement are required to enhance the formulation and administration of hTERT immunotherapy for prostate cancer. Additionally, combination therapies that target various biological components of hTERT and synergize with other treatment modalities, such as chemotherapy, radiation, or hormone therapy, may improve the prognosis for prostate cancer patients.
Survivin
Survivin, an inhibitor of apoptosis and activator of proliferation, is produced by numerous tissues during fetal development, but its expression becomes essentially nonexistent in differentiated cells[113]. However, in many human malignancies, including PCa, survivin is significantly overexpressed and its presence is correlated with tumor development, poorer prognosis for malignant illness, and medication resistance. Survivin has been identified as a modulator of antiandrogen treatment resistance in PCa[111,188,189]. Various preclinical models have shown that immunotherapeutic approaches targeting survivin can elicit T cell responses and antitumor activity[190,191]. Survivin is, therefore, a possible target for cancer treatment, particularly in immunotherapy, which aims to activate the immune system to identify and eliminate cancer cells[192,193].
Given its crucial role in tumor survival and resistance, survivin becomes a particularly pertinent immunotherapeutic target in prostate cancer. Targeting survivin, along with other tumor antigens in prostate cancer, necessitates the use of various immunotherapeutic strategies that have either been established or are currently under research. Although several of these methods have demonstrated encouraging results in clinical trials, they are not without difficulties and restrictions. Thus, further research is required to maximize the effectiveness and safety of various therapies and to determine the ideal patients for each form of treatment.
HER-2/neu, EGFR, HER-4 (Epidermal Growth Factor Family)
The ErbB receptor tyrosine kinase family, which includes cell surface proteins such as EGFR, HER 1, HER 2/neu, and HER 4, is often found to be overexpressed in various types of tumors, including PCa[110]. These proteins present promising targets for T cells or antibody-dependent immunotherapy. Specifically, overexpression of HER 2/neu in early PCa is linked to adverse clinical results, including primary recurrence and reduced overall survival[194,195]. HER 2/neu is included in the development of prostate cancer towards androgen independence. A wide array of HER 2/neu-derived proteins that are HLA class I-, class II-restricted have been identified[196]. Furthermore, mice models of various types of solid tumors have demonstrated the efficacy of active immunotherapy[197]. In preclinical models, treatments involving monoclonal anti-HER 2/neu antibodies have been investigated. In both androgen-dependent xenograft models and a combined therapy approach including HRPC xenograft models with tyrosine kinase inhibitors, administration of trastuzumab significantly inhibited the growth of established tumors[197,198]. Additionally, in a SCID mouse model, specifically engineered T lymphocytes targeting Her-2/neu successfully attacked prostate cancer (bone marrow metastases)[199].
A significant proportion of PCa cases exhibited elevated EGFR levels, which has been associated with the progression of PCa towards androgen independence. In vivo trials administering EGFR-specific monoclonal antibodies cetuximab and panitumumab have demonstrated the suppression of cancer development in multiple prostate cancer xenograft models[111,200]. Despite EGFR primarily being investigated as a target for monoclonal antibodies, several CTL epitopes have been found[111,201]. Moreover, HER4 has emerged as a potential target molecule in prostate cancer research. Both mouse xenograft models and In vitro studies have shown that HER 4 antibodies effectively slow the development of various types of prostate cancer. Additionally, concomitant radiation therapy may enhance the efficacy of HER 4-directed monoclonal antibody treatment[111,202].
HER 2/neu, EGFR, and HER-4 are prospective targets for immunotherapy in prostate cancer. However, their precise functions and effectiveness are still under investigation. Various factors, such as tumor heterogeneity, receptor dimerization, ligand availability, signaling crosstalk, immune evasion, and genetic changes, may affect both the responsiveness and resistance to targeting these receptors in prostate cancer. Therefore, further study is required to refine patient selection, therapy combination, and outcome monitoring in prostate cancer immunotherapy targeting the HER family.
N-Cadherin
N-cadherin, a protein integral to cell adhesion and migration, plays a pivotal role in various aspects of tumor progression, including the facilitation of epithelial-mesenchymal transition and the augmentation of cell movement rate. Its upregulation is a hallmark of tumor invasiveness and metastasis formation, such as pelvic lymph node infiltration and bone metastases in PCa[203,204]. Furthermore, in PCa, N-cadherin overexpression has been linked to de-differentiation, androgen deprivation, and the shift to androgen independence. Efforts to target castration-resistant PCa using mAbs have yielded promising results, significantly slowing the development of such xenografts, inhibiting invasion and metastasis, and delaying the development of androgen resistance[203,206]. Given its crucial role in tumor survival and resistance, N-cadherin emerges as a special target for prostate cancer immunotherapy. Consequently, numerous immunotherapeutic strategies have been devised or are currently under investigation to specifically target N-cadherin in prostate cancer. Although several of these methods have demonstrated encouraging results in clinical trials, they are not devoid of difficulties and restrictions. To maximize the effectiveness and safety of various therapies and to identify the most suitable patients for each treatment modality, more study is required.
Erythropoietin producing hepatocellular receptor tyrosine kinase class A2
Erythropoietin producing hepatocellular receptor tyrosine kinase class A2 (EphA2), a cell membrane-attached receptor with tyrosine kinase activity, is expressed in a wide variety of normal tissues and is notably overexpressed in numerous epithelial malignancies, including prostate cancer[203-207]. In advanced PCa, EphA2 shows promise as a potent molecule for both active and passive immunotherapy. Various HLA class I- and II-restricted proteins have been discovered, and when pulsed onto DCs, some of these peptides exhibited antitumor effects in mice models[111].
Extensive investigation into the immune-therapeutical potential of monoclonal antibodies targeting EphA2 was conducted using preclinical in vivo models. These antibodies, by effectively downregulating EphA2 on the cell surface[208,209], induce receptor internalization and disruption. Additionally, by decreasing tumor cell proliferation, promoting apoptotic activity, and reducing microvascular density, they exhibit robust antitumor effects when used alone or in combination with chemotherapy in a variety of xenograft models[111,210,211]. Cytotoxic immune-conjugates offer a highly effective way to target tumors because they quickly internalize receptors when an agonistic antibody targets them[111,212].
However, there are certain difficulties and restrictions when using EphA2 as a potent protein for prostate cancer immunotherapy. For instance, regarding the signaling pathway involved, EphA2 can exhibit both tumor-promoting and tumor-suppressive properties. EphA2 acts as a tyrosine kinase, promoting tumor growth by stimulating AKT, or functions as a pseudokinase, preventing cancer cell growth by blocking AKT activation through ephrin-A1 stimulation[211]. Therefore, in the development of EphA2-targeted therapeutics, it is crucial to consider the balance between these two mechanisms of action. Moreover, the variability in EphA2 expression and function across different prostate cancer subtypes and stages presents another problem. In distinct prostate cancer cells, EphA2 expression levels can range from high to low, or even be absent altogether, with its function shifting from proangiogenic to antiangiogenic as tumors progress[208]. This underscores the need for further research to find a more potent and effective target in prostate cancer.
Synovial sarcoma X-chromosome breakpoint protein
The family of CTAs (cancer testis antigens) known as Synovial sarcoma X-chromosome breakpoint protein (SSX) proteins is typically present in the testis and is abnormally expressed in various types of malignancies, including prostate cancer[213]. SSX1, SSX2, and SSX4 genes, involved in the typical chromosomal translocation t (X, 18) (p11, q11) seen in synovial sarcoma, encode for SSX proteins. This translocation results in the production of the SS18-SSX fusion protein, which is responsible for the pathogenesis of synovial sarcoma by joining the SS18 gene (chromosome 18) to one of the SSX genes (chromosomal X)[214]. SSX proteins, characterized by nuclear localization and limited expression in germ cells of testis or ovary lacking HLA class I, are frequently observed in tumors of various origins, particularly in advanced stages of cancer, making them a superfamily of homologous CTAs[111,215]. Due to their modest expression in healthy cells displaying HLA-class I, SSX peptides are attractive targets for T cell therapies in immunotherapy. Recent research on PCa has revealed that while SSX protein expression is absent in primary tumors, it is detected in a significant portion of metastatic PCa samples[211]. Moreover, the degree of protein homology suggests that several of the HLA class I- and II-restricted proteins represent target structures for malignancies expressing various members of the SSX family[213-216].
Epithelial cell adhesion molecule
Epithelial cell adhesion molecule (EpCAM) is commonly expressed in epithelial cells found in various tissues such as the skin, gut, and prostate. In conditions like prostate cancer, there may be instances of EpCAM overexpression or mutation, which are associated with directional growth inhibition and increased invasiveness[217,218]. Consequently, EpCAM has been identified as a promising target for immunotherapy in prostate cancer[219], a treatment strategy that harnesses the immune system to combat cancer cells. Immunotherapy relies on targeting specific molecules present on the surface of cancer cells, such as EpCAM, to stimulate the immune system's response against them. Various tools, including monoclonal antibodies, immunotoxins, and small-molecule inhibitors, can be employed for this purpose[217,218].
In both preclinical and clinical investigations, a few of these drugs have shown positive anticancer benefits. For instance, ALW-II-41-27 (a small molecule inhibitor) binds specifically to EpCAM's ATP-binding pocket, inhibiting its kinase function. In vitro and in vivo experiments have demonstrated that this substance halts the growth and invasion of prostate cancer cells[218]. Similarly, KB004, a humanized monoclonal antibody, induces the internalization and degradation of EpCAM by recognizing an epitope in its extracellular region. The presence of this antibody has been shown to slow down the development and angiogenesis of prostate cancer xenografts in mice[218]. However, utilizing EpCAM as a target for prostate cancer immunotherapy comes with certain limitations. Depending on the situation and the signaling pathway involved, EpCAM can exhibit both tumor-promoting and tumor-suppressive functions. For example, it can act as a tyrosine kinase, promoting tumor growth through the activation of AKT, or as a pseudokinase, inhibiting AKT activation via ligand stimulation[217]. Therefore, when developing EpCAM-targeted medicines, it is crucial to carefully balance these opposing mechanisms. Furthermore, the variability in EpCAM expression and activity across different prostate cancer subtypes poses another challenge. EpCAM expression levels can vary from high to low, or even be missing in different prostate cancer cells, and its function can change from proangiogenic to antiangiogenic with tumor progression[218]. Therefore, the ideal timing and dosage of EpCAM-targeted therapy should be carefully evaluated based on the specific characteristics of each individual case.
Receptor-interacting protein kinase 2
Receptor-interacting protein kinase 2 (RIPK2) is a protein crucial for innate immunity and inflammation. Additionally, it plays a role in the advancement and metastasis of prostate cancer by maintaining c-Myc, a protein that promotes tumor development and invasiveness[221]. Therefore, interventions targeting RIPK2 to degrade c-Myc, such as medicines or gene editing techniques, may hold promise in preventing prostate cancer[220,221]. Some research suggests that RIPK2 inhibitors have shown beneficial effects in both preclinical and clinical trials. For instance, the small molecule inhibitor ALW-II-41-27 binds to the ATP-binding site of RIPK2, impeding its kinase activity. Studies conducted in vitro and in vivo have demonstrated its efficacy in inhibiting the proliferation and invasion of prostate cancer cells[220]. Another approach involves KB004, a humanized monoclonal antibody that induces internalization and degradation of RIPK2 by recognizing an epitope on its extracellular domain. In mouse models with prostate cancer xenografts, the presence of this antibody has been demonstrated to slow down tumor development and angiogenesis.
However, there are certain difficulties and restrictions when using RIPK2 as a target for prostate cancer immunotherapy. Depending on the context and the signaling pathway involved, RIPK2 can serve both oncogenic and tumor-suppressive purposes. It can either activate AKT as a tyrosine kinase or inhibit its activation by ligand stimulation as a pseudokinase[221,222]. Therefore, when developing RIPK2-targeted therapeutics, the balance between these two mechanisms of action should be carefully considered.
Another obstacle is the variability in RIPK2 expression and function across various subtypes and stages of prostate cancer. In various prostate cancer cells, RIPK2 expression levels can range from high to low, or even absent, and its function can change from proangiogenic to antiangiogenic as tumors progress[220]. As a result, RIPK2-targeted medicines should be carefully tailored in terms of timing and dosage to accommodate the unique characteristics of each patient.
MAGE
Melanoma antigen gene protein-A11 (MAGE-11), a member of the MAGE family of cancer germ-line antigens, interacts with the AR NH(2)-terminal FXXLF motif, thereby promoting androgen receptor (AR) transcriptional activity[223]. In addition to activating genes necessary for male sex differentiation, high-affinity androgen binding to the androgen receptor (AR) accelerates the onset and progression of prostate cancer[224]. Interactions between human AR transcriptional activity and coregulatory proteins involve the coactivator melanoma antigen-A11 (MAGE-A11), which is primate-specific and promotes AR transcriptional activity, thereby contributing to the progression of prostate cancer to castration-resistant/recurrent prostate cancer (CRPC)[225]. Notably, MAGE-A11's involvement in modulating hormonal signals in prostate cancer sets it apart from other type I MAGEs. The binding of MAGE-A11 to the N-terminal FXXLF motif of the androgen receptor (AR) facilitates SRC/p160 coactivator binding[226]. The phosphorylation and ubiquitination of MAGE-A11, regulated by epidermal growth factor (EGF), have also been found to enhance AR transcriptional activity[227].
ROLE OF IMMUNE CELLS IN ADVANCED PROSTATE CANCER
Extensive research has delved into the complicated dynamics of immune cells in advanced prostate cancer. Immune cells serve as the body's natural defense mechanism against various intruders, including bacteria, viruses, and cancer cells. However, in some cases, immune cells can promote tumor growth and metastasis by creating a favorable environment for cancer cells to proliferate and spread. Based on their immune infiltration patterns, cancers are immunologically divided into two categories: cold tumors [Figure 3] and hot tumors. Hot tumors are characterized by high filtration of T cells and cytotoxic T lymphocytes (CTLs), primarily due to increased tumor mutational burden, and an increase in peptides, which activate checkpoint amino acids[228].
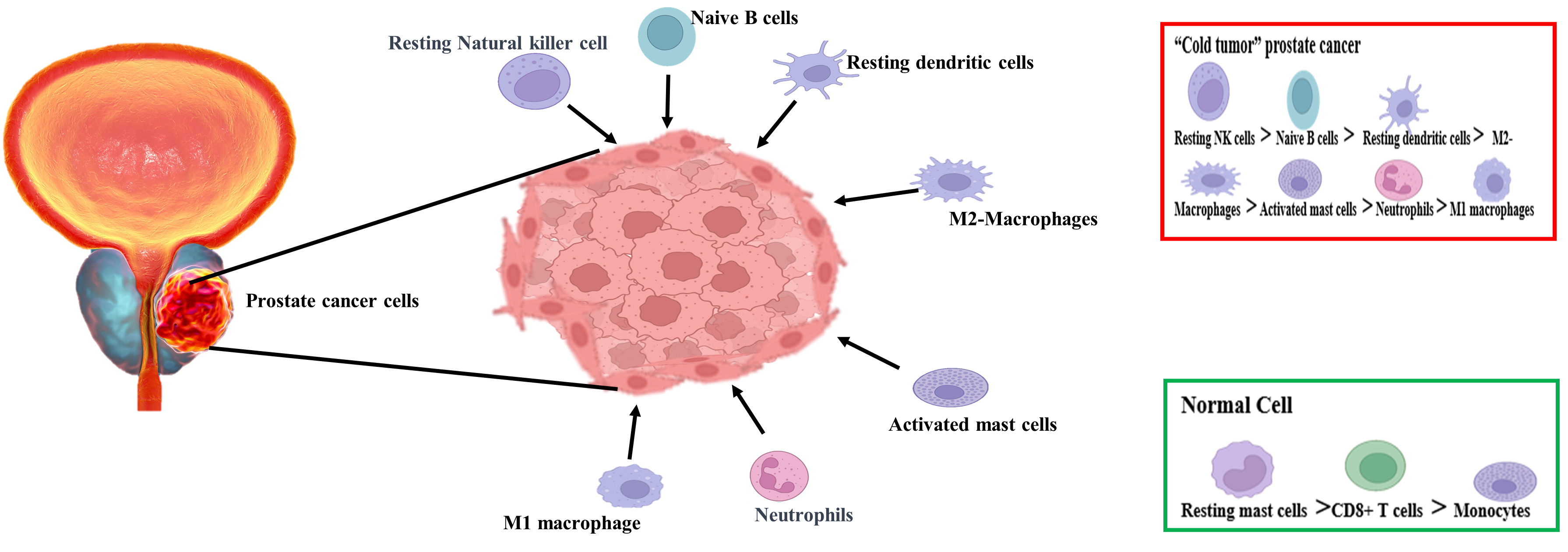
Figure 3. Difference in infiltrating proportions of immune cells in the normal and prostatic cells. In prostate cancer, the percentage of infiltration of resting NK cells increased the most, whereas the percentage of infiltration of resting mast cells decreased the most. In normal tissues, CD8+ T cells had the strongest infiltrating correlation with monocytes, while activated NK cells and naive B cells were the highest in prostate cancer tissues.
Conversely, cold tumors exhibit low mutational burden, poor antigen expression, the presence of tumor-associated macrophages (TAM) polarized towards a pro-tumor (M2-like phenotype), and exhausted CTLs within the tumor or their absence at the tumor margins. PCa can be viewed as a tumor with a cold immune system. The tumor microenvironment (TME), comprising immune cells, significantly influences cancer development and the response to immunotherapy[228,229].
In comparison to benign nodular hyperplasia of the prostate, prostatic adenocarcinomas have been found to have a low density of immune cells [Figure 3][230]. The activity of antitumor CD8+ T cells is inhibited and slowed down by the upregulation of nitric oxide synthase and the secretion of indoleamine 2,3-dioxygenase (IDO) by myeloid-derived suppressor cells (MDSCs). Additionally, there is a high presence of regulatory T cells (Tregs) compared to other cancers, along with other immune-suppressive cells like neutrophils or M2 TAM, both of which are associated with poor survival outcomes. The secretion of specific substances within the TME, such as TGF-β and CXCR2, further supports this immunosuppressive milieu. Promisingly, the recent clinical trial (NCT03473925) suggests that inhibiting CXCR2 may be useful for enhancing immunotherapy[231-233].
Due to the heightened blood flow in red bone marrow, there is an increased interaction between stromal cells and tumor cells, and stomal cells release growth factors, angiogenic factors, and bone-resorbing factors that promote tumor growth, with bone being the preferential site of nearly 90% of PCa metastases. The formation and development of PCa bone metastases depend on the tumor immune microenvironment[234-236]. In an in vivo mouse model, tumor cells disseminate and produce IL-6, which interacts with tumor-associated macrophages (TAMs) and promotes tumor cell proliferation and angiogenesis in bone locations. Additionally, considerable amounts of TGF-β are present in bone metastases, where they cause CD4+ helper cells to transform into Tregs, contributing to the ineffectiveness of immunotherapies in metastatic castration-resistant prostate cancer (mCRPC)[237]. Focusing on the release of different types of factors at preferred metastatic locations may therefore present a potential strategy. Additionally, an increased ratio of M2-like to M1-like TAMs was linked to lower survival in prostate cancer patients. The ratio of M2-like to M1-like TAMs was higher in metastatic prostate cancer than in localized prostate cancer[238]. Pro-tumoral TAMs, sometimes referred to as M2-like TAMs, can accelerate angiogenesis, invasion, and immunosuppression to aid in the development and metastatic growth of tumors. Matrix metalloproteinases (MMPs), interleukin-10 (IL-10), vascular endothelial growth factor (VEGF), and transforming growth factor-β (TGF-β) are among the substances released by M2-like TAMs, which promote tumor cell proliferation, survival, and migration[239]. Antiangiogenic treatments have demonstrated significant advantages in the treatment of various cancer types, yet their efficacy in the case of pancreatic cancer is only modest. Among others, prostate cancer can produce matrix metalloproteinases, vascular endothelial growth factor (VEGF), transforming growth factor β (TGFβ), and cyclooxygenase-2 (COX-2). The microenvironment surrounding prostate cancer plays a crucial role in its onset and development. Micro-vessel density, a measurement of prostate cancer angiogenesis, has been shown to be a predictor of metastasis and survival, and therefore, targeting angiogenesis has been the subject of several clinical investigations and debates. Moreover, the prognostic potential of angiogenic activity measurement holds great promise[240].
PCa can invade lymph nodes, establishing a pre-metastatic microenvironment and altering their structure and immune response. In actuality, an immunosuppressive microenvironment is created. In PCa subjects with pelvic lymph nodes, MDSCs, which comprise monocytes and granulocytes, express immune-suppressive peptides such as programmed cell death ligand 1/2 (PD-L1/L2)[241,242]. These MDSCs inhibit the growth of CD8+ T lymphocytes gathered in pelvic lymph nodes that show immunological checkpoint proteins due to their immunosuppressive action. Since there are fewer antigen-presenting DCs in the paracortical region, the responsiveness of antitumor T cells may change[241]. By regulating T cells, tumor-derived extracellular vesicles may be responsible for shaping a pre-metastatic niche in lymph nodes. When considered as a whole, tumor microenvironment (TME) cells at sites of prostate cancer metastatic development promote immune evasion and tumor development[243,244]. Immunotherapies can potentially remodel the TME to target prostatic metastases. Additionally, patients with advanced prostate cancer may have higher levels of MDSCs in their blood and tissues, potentially accelerating disease progression and complicating treatment efforts[245].
IMMUNE CHECKPOINT INHIBITORS AND PROSTATE CANCER
Immune checkpoint inhibitors are a form of immune therapy that aids the immune system in identifying and combating cancer cells. These inhibitors work by blocking proteins that prevent the immune system from attacking cancer cells. By inhibiting these proteins, immune checkpoint inhibitors can shrink tumors or slow down their growth, enhancing the immune response against cancer cells[246]. Despite their success in treating various cancers, the application of immune checkpoint inhibitors to prostate cancer has proven challenging and has yielded limited success. Prostate tumors typically have low mutation rates, low PD-L1 expression (a protein targeted by immune checkpoint inhibitors), and an increment of immunosuppressive elements in the tumor environment, making them unresponsive to these inhibitors[10]. Recent research, however, has indicated positive outcomes with immune checkpoint inhibitors (ICIs) and their combinations in specific patient groups with high PD-L1 expression, mutations in CDK12, elevated mutational pressure in tumors, and tumors displaying more mismatch repair deficiency (dMMR) and microsatellite instability (MSI) levels[11,247]. Immune checkpoint inhibitors are thus restricted to certain prostate cancer subtypes that have particular genetic or molecular characteristics that render them more recognizable or susceptible to the immune system [Figure 4]. Among these subtypes are:

Figure 4. Overview of how immune checkpoint inhibitors work in the context of prostate cancer. PD-L1 binds to PD-1, preventing T cells from killing tumor cells; blocking PD-L1 or PD-1 allows T cells to kill tumor cells. MSI-H (microsatellite instability) or MMR (mutational changes in the mismatch repair)-deficient prostate cancer cells, increased PD-L1 expression, increased tumor mutational burden (TMB), and CDK12 mutations contribute to a favorable response to immune checkpoint inhibitors. In some patients with MSI-H or MMR-deficient prostate cancer, ICI drugs that target PD-1 or PD-L1, such as pembrolizumab and nivolumab, have been proven to be effective.
1. Increased microsatellite instability (MSI-H) or mutational changes in the mismatch repair (MMR) genes in prostate cancer: Uncommon prostate cancer subtypes with an increment in mutation rate and the ability to create aberrant proteins that the immune system may mistake for foreign substances. In some patients with MSI-H or MMR-deficient prostate cancer, ICI drugs that target PD-1 or PD-L1, such as pembrolizumab and nivolumab, have been proven to be effective[246,248,249].
2. Prostate tumors with high PD-L1 expression: Some cancer cells express a protein that can attach to the immune cells of PD-1 receptor and stop those cells from attacking the cancer cells. By blocking this interaction, ICIs that target PD-1 and PD-L1 can activate the body's defenses against cancer cells. According to certain research, prostate tumors with more PD-L1 display may respond to ICIs more favorably than those with low PD-L13 expression[246,248].
3. High tumor mutational burden (TMB) prostate tumors: This is a measurement of the quantity of DNA mutations present in cancer cells. More aberrant proteins that the immune system may identify as foreign can result from a high TMB. Prostate tumors with high TMB may respond better to ICIs that target the PD-1 or PD-L1 proteins, compared to prostate cancers with low TMB[246,248,250].
4. Prostate tumors with CDK12 mutations: Transcription and DNA repair are the major role played by this gene. Genomic instability and a rise in the development of neoantigens—new or altered peptides identified as outsider by the immune system - can result from mutations in CDK12. Prostate tumors with CDK12 mutations may respond better to immune system checkpoint medications focused on PD-1 or PD-L1 than those without[246,248].
Overview of clinical trials involving immune checkpoint inhibitors in prostate cancer
Sipuleucel-T, an active cellular immunotherapy, demonstrated the clinical significance of the immune system in advanced prostate cancer (PCa). In a phase III trial, asymptomatic or minimally symptomatic metastatic castration-resistant PCa (mCRPC) patients receiving sipuleucel-T had a 22% lower risk of death compared to the placebo group (HR 0.78, 95%CI: 0.61-0.98), leading to a 4.1-month improvement in median overall survival. This underscores the potential of immune system modulation to benefit and improve outcomes in mCRPC[110]. In patients (CA184-095) with mildly symptomatic mCRPC who had not received chemotherapy, high-dose ipilimumab (10 mg/kg) monotherapy did not result in a better median OS compared to the placebo (28.7 months vs. 29.7 months; HR = 1.11, 95%CI: 26.1-34.2 months,
Completed Phase 3 clinical trials of immune checkpoint inhibitors in Metastatic castration-resistant prostate cancer (mCRPC)
| Trial name/NCT ID | Methodologies | Number of patients enrolled | Trial type | Significant outcome |
| CA184-095/NCT01057810 | Ipilimumab verses placebo | 837 | Phase 3 | Median OS 28.7 vs. 29.7 months. No improvement in OS (Overall survival) with ipilimumab[251] |
| CA184-43/NCT00861614 | Ipilimumab vs. placebo following radiotherapy | 799 | Phase 3 | Median OS 11, 2 months vs. 10, 0 months[252] |
| KEYNOTE-641/CT03834493 | Pembrolizumab + enzalutamide vs. placebo + enzalutamide in mCRPC | 1244 | Phase 3 | Primary endpoints were not met[253] |
| KEYNOTE-010/NCT03834519 | Pembrolizumab + olaparib vs. NHA in mCRPC | 793 | Phase 3 | Median OS with Pembrolizumab + Olaparib was 15.8 months (95%CI: 14.6-17.0) compared to 14.6 months (95%CI: 12.6-17.3) in the control arm. The HR was 0.94 (95%CI: 0.77-1.14) with a P-value of 0.26[254] |
| KEYNOTE-921/NCT03834506 | Pembrolizumab + docetaxel vs. docetaxel in mCRPC | 1030 | Phase 3 | Median OS with Pembrolizumab + Docetaxel was 19.6 months (95%CI: 18.2 to 20.9) compared to 19.0 months (95%CI: 17.9 to 20.9) with Docetaxel alone. The HR was 0.92 (95%CI: 0.78-1.09) with a P-value of 0.1677[255]. |
| IMbassador250/NCT03016312 | Atezolizumab + enzalutamide vs. placebo + enzalutamide in mCRPC | 772 | Phase 3 | Median OS with atezolizumab + enzalutamide was 15.2 months (95%CI: 14.0-17.0) compared to 16.6 months (95%CI: 14.7-18.4) in the control group. The HR was 1.12 (95%CI: 0.91-1.37) with a P-value of 0.28[256]. |
| CheckMate-7DX /NCT04100018 | Nivolumab + docetaxel vs. Placebo + docetaxel in mCRPC | 984 | Phase 3 | Primary endpoints were not met[257,258]. |
In the KEYNOTE-028 phase Ib study, pembrolizumab showed a 17.4% objective response rate (ORR) in 23 heavily treated mCRPC patients with measurable disease and ≥ 1% PD-L1 expression. The response included a partial response (PR) in 4 patients, with 3 experiencing a parallel biochemical response. The trial had a favorable side effect profile[259]. Subsequently, pembrolizumab was studied in the KEYNOTE-199 trial across three mCRPC cohorts. Cohort 1 had PD-L1-positive tumors, cohort 2 had PD-L1-negative tumors, and cohort 3 had non-measurable bone metastatic disease. Median overall survival (OS) was 9.5, 7.9, and 14.1 months for cohorts 1, 2, and 3, respectively. Confirmed PSA response rates were 6%, 8%, and 2% in the respective cohorts. The observed ORR was modest (about 5%), with a median response duration of 16.8 months. Notably, outcomes were similar regardless of PD-L1 status, and no clear relationship was found between pembrolizumab responses and DNA damage repair gene mutations[260,261].
These are a few immune checkpoint inhibitors applied to cure prostate cancer. For the majority of prostate cancer patients, these medications are ineffective, and they are not approved for all patients with the illness. Moreover, not all patients with these subtypes will react to immunotherapy, and the reaction to immune checkpoint inhibitors is still uncertain and variable. The first immunotherapeutic medication to be licensed by the FDA for the treatment of metastatic castrate-resistant prostate cancer (mCRPC) that is without any symptoms and with some poor symptoms is sipuleucel-T (Provenge), which increases the overall survival ratio of the subjects. Limited responses have been observed in mCRPC despite numerous clinical trials investigating ICIs and combinations of these with different medications. Thus far, ICIs binding with different molecules, like DNA damage molecules, have only been clinically beneficial for a limited subgroup of subjects by CDK12 mutations, MMR deficiency, more MSI, and these conditions[1]. To overcome resistance and enhance the effectiveness of immunotherapy in treating prostate cancer, further research is required to uncover biomarkers and assess the efficacy and toxicity of ICIs.
REASONS FOR IMMUNOTHERAPY FAILURE IN PROSTATE CANCER
Some malignancies are more resistant to immunotherapy than others, and it may not always work. One malignancy that has had a poor response to immunotherapy, particularly in advanced stages, is prostate cancer. Potential causes for immunotherapy failure in prostate cancer include:
1. Absence of tumor inflammation: Neoantigens, which are novel or changed peptides recognized by the immune system as foreign molecules, are infrequent in prostate cancer cells. Consequently, immune cells are less drawn to prostate cancer, and there is little tumor inflammation. Increased tumor inflammation improves the efficacy of immunotherapy by providing a more conducive environment for immune cells to target cancer cells[262,263].
2. Low quantities of antigens, which are chemicals that can elicit an immune response, are present in prostate cancer cells. This makes it challenging for the immune system to identify and combat them[233,264].
3. A microenvironment that inhibits or evades the immune system is created by prostate cancer, which produces immunosuppressive chemicals, attracts regulatory T cells, and expresses checkpoint ligands, among other things[233,264].
4. Due to its considerable heterogeneity, prostate cancer might respond differently to immunotherapy and develop resistance to it. The cancer comprises several clones and subtypes with varying genetic and molecular properties[233,264].
5. Due to the low incidence of DNA repair errors, there are not many mutations in prostate cancer that could increase its susceptibility to checkpoint inhibitors[233].
6. The intricate relationship between androgen receptor and prostate cancer hinges on testosterone, which is crucial for both development and survival. Testosterone can affect the expression and function of immune cells and molecules. However, despite this understanding, the currently available immunotherapies licensed for prostate cancer (sipuleucel-T and pembrolizumab) have only demonstrated minor improvements in a limited subset of patients, underscoring the inadequacy of effective immunotherapies for prostate cancer[264-266].
7. Immune checkpoint inhibitors that stop the immune cells with inhibitory receptors, such as PD-1 and CTLA-4, protect the cancer cells from the attack or inhibitory act of immune cells. Checkpoint blockade has demonstrated efficacy in treating certain cancers such as lung and melanoma cancer, but its effectiveness in prostate cancer has been limited[267]. Additionally, melanoma of unknown primary (MUP) remains biologically ill-defined, as compared to the classical melanoma of known primary (MKP). Recent research has revealed that patients with MUP sites seem to present better outcomes compared to those with stage-matched MKP, probably due to higher immunogenicity as reflected in the immunologically mediated primary site regression. MUP patients on immunotherapy probably display better outcomes compared to the MKP subset[268]. Researchers investigated the possible reasons for this resistance and found that it is driven by immune cells called macrophages. They showed that macrophages in prostate tumors can produce a protein called IL-23, which can activate a signaling pathway called STAT3 in tumor cells. STAT3 can then induce the efficacy of genes that increase tumor survival and inhibit checkpoint inhibitors. In preclinical models of prostate cancer, the researchers also demonstrated that inhibiting IL-23 or STAT3 could improve the effectiveness of checkpoint blockade[267].
The influence of co-morbidity on the response of immunotherapy in prostate cancer is one of the causes of immunotherapy failure. In a study conducted in 2020, researchers found that 11% of the patients included had a history of diabetes. Further investigations into a national sample revealed that 13% of prostate cancer patients had this condition. Additionally, hypertension was mentioned by 56% of patients who underwent radical prostatectomy for prostate cancer. Within this sample, 18 out of 42 patients were diagnosed with hypertension. Nonetheless, a national sample of prostate cancer patients showed that 30.5% of them had at least one co-morbidity[268,269]. Contrary to other studies, such as the one by Edwards et al., which predicted co-morbidity conditions included in the Charlson Co-Morbidity Index, excluding hypertension, our higher overall rates of co-morbidity may be due to the inclusion of hypertension[270,271]. Considering earlier research showing that men undergoing radical prostatectomy have a higher chance of biochemical recurrence post-treatment, the absence of hypertension in studies analyzing the prognosis of prostate cancer may be a noteworthy oversight[272,273].
CURRENT STATUS OF GENE THERAPY IN PROSTATE CANCER
After the discovery of the DNA helical structure, there was a global technological explosion, leading to the development of numerous cutting-edge technologies that are currently being implemented in therapeutic settings. Several molecular approaches that aid in editing DNA codes and post-transcriptional mRNA modification have come over the past few decades. Delivering certain genetic material to alter a gene product's encoding or a tissue's biological characteristics in order to treat a variety of illnesses is known as gene therapy[274]. The drawbacks of using peptides in recombinant therapeutics, including limited bioavailability, instability, extreme toxicity, clearance rates, and expensive production costs, are addressed via gene therapy[275]. Gene treatments work through a variety of techniques, such as administration of newly identified genes to cure a disease, knocking down or deactivating problematic genes, and replacing malfunctioning genes with therapeutic ones[276]. In order to transport foreign genetic material into the host organ, gene therapy employs RNA and DNA by using different transferring vehicles/vectors. In in vivo gene therapy, genetic material is delivered directly into target organs, while ex vivo gene therapy involves modifying host cells outside the body before reintroducing them. The goals of gene therapy are to restrict the activity of the affected gene, enhance the presence of modifier disease genes, or supply a functioning gene copy of the destroyed gene[277,278] [Figure 5].
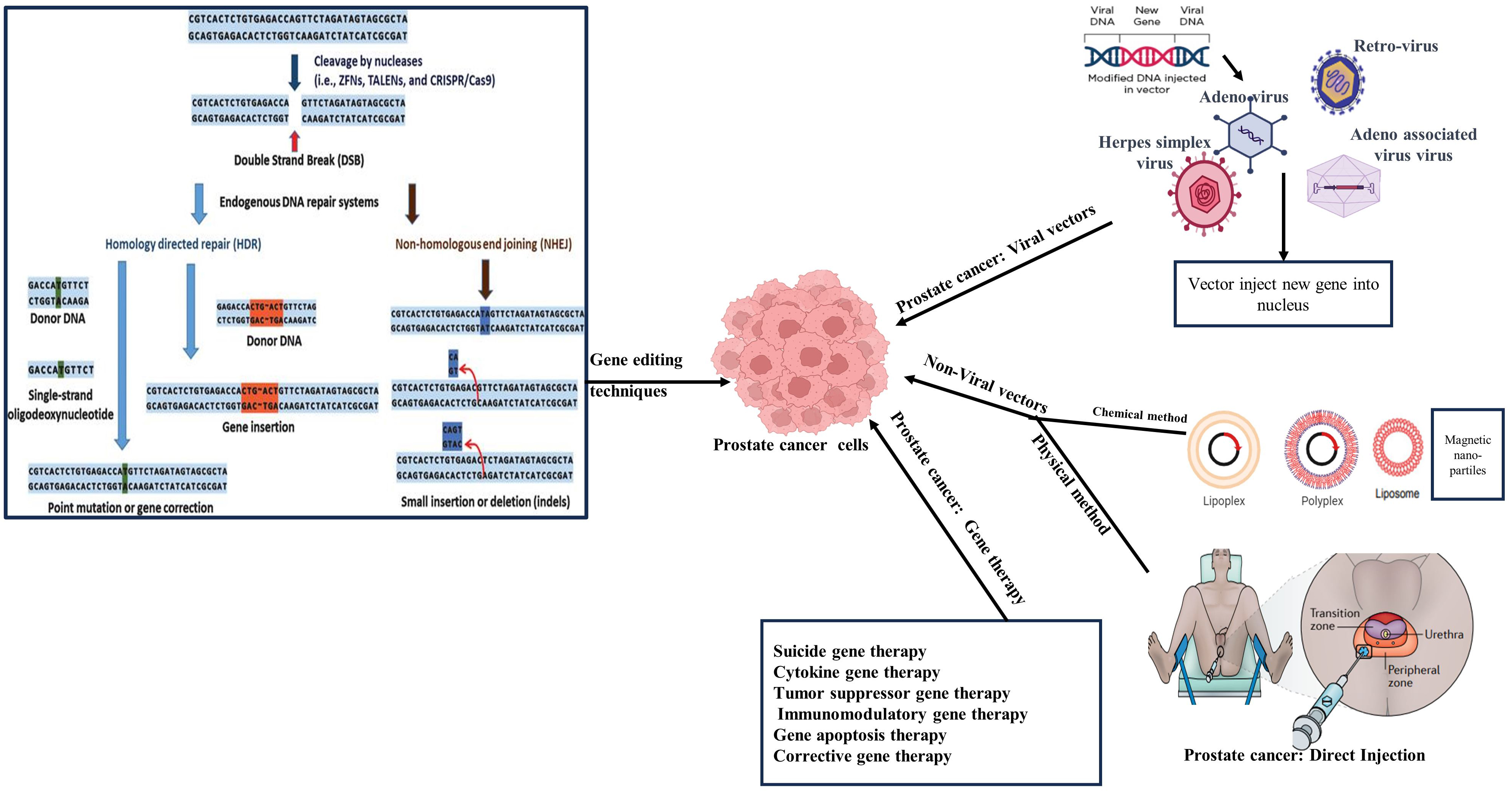
Figure 5. Schematic overview of gene therapy in Prostate cancer. Based on their structural differences, there are currently four types of nucleases that edit genes in prostate cancer: base editors, zinc-finger nucleases (ZFN), transcription activator-like effector nucleases (TALENs), and CRISPR-associated nucleases (CRISPR/Cas-9). Viral vectors including adenovirus (Ad), adeno-associated virus (AAV), herpes simplex virus (HSV), and retroviruses (γ-retroviruses, lentiviruses) play a significant role in the transport of genetic materials. Genetic materials are introduced through cell membranes via physical methods such as direct injection and chemical methods such as lipoplex, polyplex, and magnetic nanoparticles. Gene therapies can be classified according to the type of transporter used, which can be viral and non-viral vectors. These therapies include suicide gene therapy, cytokine gene therapy, tumor suppressor gene therapy, immunomodulatory gene therapy, gene apoptosis therapy, and corrective gene thera.
Gene editing techniques
Gene editing is a technology that makes it possible to change an organism's or cell's DNA sequence. Gene editing can be used to improve features, develop medicines, create animal models, and rectify genetic flaws, among other things. Nucleases, which are enzymes that can cut DNA at certain places, are essential to the process of gene editing. Based on their structural differences, there are currently four types of nucleases that edit genes in prostate cancer: base editors, zinc-finger nucleases (ZFN), transcription activator-like effector nucleases (TALENs), and CRISPR-associated nucleases (CRISPR /Cas-9) [Figure 5].
Base editors
Base editors are a subset of nucleases used in gene editing that can transform one base into another without inducing double-strand breaks (DSBs) in the DNA. Base editors consist of an adenine-to-inosine or cytosine-to-uracil deaminase enzyme coupled with a Cas9 variant. The Cas9 variant can only bind to or nick DNA depending on whether it is catalytically inactive (dCas9) or nickase (nCas9). The guide RNA binding site is surrounded by a small window where the deaminase enzyme can work on the target base. The cellular DNA repair machinery then corrects the resultant base alterations. Base editors are useful for introducing precise base alterations or correcting point mutations in genes linked to prostate cancer, such as androgen receptors (AR)[279,280].
ZINC finger nucleases
Zinc finger nucleases (ZFNs) are synthetic nucleases made up of a pair of domains: a FokI nuclease domain that breaks the DNA and a zinc finger domain that detects and binds to a particular DNA sequence. By manipulating the zinc finger domain, which has three to six zinc finger repeats that individually recognize three to eighteen base pairs, ZFNs can be engineered to target desired DNA sequence effectively[269,281]. Double-strand breaks (DSBs) in DNA can be caused by ZFNs and corrected by non-homologous end joining (NHEJ) or homology-directed repair (HDR), which involve either insertion and disruption of gene. Plasmid-based techniques circumvent the drawbacks of viral administration, including immunogenicity, toxicity, and insertional mutagenesis, and can transfer ZFNs to the intended cells. Certain disadvantages of ZFNs include the potential for off-target breaks, which can result in the destruction of cells, and the random integration of DNA donors. Additionally, when ZFNs fail to precisely target or focus on a specific spot, the likelihood of off-target break increases. ZFNs have been employed in the modification of prostate cancer-related genes such as AR, PTEN, and TMPRSS2-ERG[282,283].
CRISPR-associated nucleases (CRISPR/Cas9)
Bacteria possess an adaptive immune system called CRISPR, which is heritable. This system helps the bacteria guard against re-infection by remembering past viral infections. In contrast to the immune system of human beings, CRISPR is inherited by the subsequent types or growth stages of bacteria, protecting the colony from further viral infections. The functionality of the CRISPR-linked immune system requires the binding of invader DNA (virus, plasmid) with the bacterial genome[284]. CRISPR/Cas9 comprises Cas9 endonuclease and short noncoding guide RNA (gRNA) consisting of two parts: helper trans-activating RNA (tracrRNA) and CRISPR RNA (crRNA) that targets specific sequences. Through base pairing between the target sequence and the crRNA sequence, the gRNA (guide RNA) unit directs Cas9 to a particular genomic location[285]. CRISPR/Cas9 technology has been employed to edit genes linked to prostate cancer, including AR, PTEN, TMPRSS2-ERG, BRCA2, and CDK12[280].
Transcription activator-like effector nucleases
Transcription activator-like effector nucleases (TALENs) are synthetic nucleases created by fusing the Fok I endonuclease with a DNA binding domain, resulting in a non-specific nuclease domain that facilitates targeted cleavage by TALE effectors. The DNA-binding domain consists of a repetitive unit of 33-35 conserved peptides domain with the exception of locations 12 and 13 that are variable and strongly correlated with particular nucleotide recognition. The DNA cleavage domain of FokI endonuclease is non-specific. To function as a dimer, the FokI domain requires two constructs with distinct DNA binding domains for sites in the target genome. Improved activity depends on the quantity of peptides between the FokI cleavage domain and the TALE DNA-binding domain. TALENs are employed to induce DSBs in order to initiate DNA repair processes and thus facilitate genome modification[281,286,287]. Prostate cancer genes such as AR, PTEN, and PDL1 have been edited using TALENs[280].
Gene delivery methods
A vector serves as the vehicle used to transfer the desired gene. Gene products can be delivered using viral vectors, nonviral vectors, as well as chemical and physical vectors [Figure 5]. The selection of appropriate vectors is an important initial step towards achieving success in gene therapy.
Viral vectors for gene delivery
The first popular vectors for gene delivery to specific tissues were viruses. Viral surface proteins interact with host receptors to trigger endocytosis. Upon entry, viruses discharge their genome into the nucleus to facilitate gene expression. The most significant viral vectors include adenovirus (Ad), adeno-associated virus (AAV), herpes simplex virus (HSV), and retroviruses (γ-retroviruses, lentiviruses)[288-290].
Nonviral vectors
Biocompatible materials such as lipid molecules, naked DNA, chromosomes, plasmids, and conjugate complexes, along with positively charged molecules, are less likely to induce immune responses in the body compared to nonviral vectors or delivery methods for gene therapy[284]. Genetic materials are introduced through cell membranes via a physical process known as nonviral gene delivery. Neon injection, ballistic DNA injection, sonoporation, photoporation, magnetofection, and hydroporation are examples of physical gene delivery techniques. The direct infusion of genomic content using a needle is referred to as needle injection. Magnetofection is the process of concentrating DNA or RNA into target cells with the assistance of magnetic molecules[291]. Magnetic nanoparticles are nanosized particles that are susceptible to external magnetic fields and possess magnetic characteristics. By attaching magnetic nanoparticles to gene carriers (plasmids, siRNA, CRISPR/Cas9, or miRNA) and using a magnetic field to guide them to the tumor site, therapeutic genes can be delivered into the target cells. Because magnetic nanoparticles can pass through some of the obstacles in the TME - such as the extracellular matrix, blood flow, and interstitial pressure - they can enhance the efficiency and specificity of gene transport[292].
Gene therapy for prostate cancer treatment
Cancer arises when the regular processes of cell growth and programmed cell death, known as apoptosis, are disturbed. Advancements in cancer therapy require innovative drugs with unique mechanisms of action, multiple methods of inducing cell death, and compatibility with existing treatments. These criteria are also applicable to gene-based treatments, representing a promising avenue for advancing cancer therapy[269].
Gene therapy studies using viral vectors
Several gene therapy strategies employing viral vectors, such as suicide gene therapy involving the addition of genes that alter enzymes and chemical molecules to generate harmful compounds, cytokine gene therapy, and tumor suppressor gene therapy, are being developed or under investigation for the management of prostate cancer[269] [Figure 5] (as following explained in 9th section).
Gene therapy studies using nonviral vectors
Prostate cancer treatment employs diverse gene therapy methods, including gene apoptosis therapy that substitutes the altered gene with one that regulates the death of cancer cells. Corrective gene therapy utilizes tumor suppressor genes to regain regular cell developmental growth, while immunomodulatory gene therapy activates the immune system with specific genes. Additionally, suicide gene therapy introduces genes that cause alterations in chemical compounds or enzymes, forming toxic substances. These strategies represent the varied approaches used in prostate cancer gene therapy[293] [Figure 5]. In addition, by employing targeting ligands that can identify certain receptors in the target tissue, the targeting capacity of nonviral vectors can be increased[293], as explained in the following 9th section.
POSSIBILITY OF SUCCESS OF GENE THERAPY IN PROSTATE CANCER
Numerous variables, including the virus type, delivery method, dosage, target tissue, immune response, and possible side effects, affect the safety and effectiveness of viral vectors. Regarding transfection efficiency, stability, specificity, immunogenicity, toxicity, and insertional mutagenesis, various viral vectors have different benefits and drawbacks. For instance, adenoviruses have the ability to transfer large genes and infect both dividing and non-dividing cells. However, they can cause powerful immune reactions and temporary expression. Conversely, retroviruses carry the risk of oncogenesis and insertional mutagenesis even though they can integrate into the host genome and produce stable, long-term expression. To guarantee the safety and effectiveness of each gene therapy strategy, it is essential to select the best viral vector[294]. The creation and design of the viral vectors, guide RNAs, or gene carriers determines the specificity and delivery of the therapeutic genes. The objective is to precisely and specifically alter or express a gene in tumor cells while preventing side effects or harm to healthy cells. By modifying their DNA-binding domains or guide RNAs, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9), zinc finger nucleases (ZFNs), and transcription activator-like effector nucleases (TALENs), for instance, can be made to target any desired DNA sequence[269,294]. They must, however, also be thoroughly examined for specificity and precision because they might result in off-target breaks or mutations that compromise the integrity or functionality of the genome. Similar modifications or optimizations are required for viral vectors or gene carriers in order to increase their particularity and efficacy of delivery to tumor location and to get past some of the obstacles present in the tumor microenvironment (such as extracellular-matrix, interstitial pressure, and blood flow)[295]. Interplay between the host immune system and the viral vectors or therapeutic genes determines the immunological response and resistance of tumor cells. Certain gene therapy techniques, such as CAR T cell (chimeric antigen receptor T cell) treatment and cytokine gene therapy, seek to activate the immune system to identify and combat tumor cells. These methods can exert strong antitumor effects, but they must also avoid triggering excessive autoimmune reactions or inflammation that could harm healthy tissues[288]. However, alternative gene therapy strategies - such as tumor suppressor or suicide gene therapy - strike against the immune system in an effort to prevent the viral vectors or therapeutic genes from being cleared out or rejected. High transfection efficiency and robust therapeutic gene expression can be attained with these methods, but immunosuppression or tolerance that could impair antitumor immunity must be avoided. Furthermore, tumor cells may acquire resistance mechanisms, such as mutation, epigenetic change, signaling crosstalk, or microenvironmental adaptability, to evade immune attack or gene therapy-induced cell death[299]. The molecular and genetic features of each patient's tumor, in addition to their clinical stage and condition, determine the suitable candidates for each treatment type and the optimal approach to combining them. Because prostate cancer is a diverse illness with several subtypes and clones with unique genetic and molecular characteristics, gene therapy response and resistance can vary. Thus, biomarker-based customized medicine is required to determine which individuals are most suited for each kind of gene therapy strategy. Patients with DNA repair abnormalities may benefit from CRISPR/Cas9 gene therapy; those with high levels of HER2 expression may benefit from trastuzumab gene therapy; and those with high PD-L1 show benefit from CAR T cell therapy[293,296]. The effectiveness of gene therapy must also be enhanced to overcome resistance, which calls for combination therapy based on synergistic effects. For instance, gene therapy in combination with immunotherapy, chemotherapy, radiation therapy, or hormone therapy may produce superior results compared to gene therapy used alone[295,297].
POTENTIAL VIRAL AND NONVIRAL GENE THERAPIES IN PROSTATE CANCER
The type of genetic material supplied, the amount of genetic material administered, and the administration method all influence the choice of vector for gene transport. Viruses as carriers offer effective transfection, long-lasting gene expression, and protection against gene deterioration; however, they can also be highly poisonous, immunogenic, have poor targeting capacity, and are very expensive. Nonviral vectors, conversely, do not elicit unfavorable immune responses and are comparatively less toxic, easy to manufacture, and capable of transmitting huge amounts of genetic material. Nonetheless, they come with their own set of limitations, such as high susceptibility to intracellular and extracellular barriers, reduced transfection capacity, and significantly lower levels of transgenic expression. Overall, every type of vector, viral and nonviral, has specific benefits and drawbacks. Nevertheless, they can be modified chemically and physically by attaching targeting compounds, additional proteins, and peptides in the form of promoters to enhance gene transport[298-301].
Potent viral gene therapies in prostate cancer
In gene therapy, viral vectors are utilized to deliver genetic materials. Gene therapy approaches that have been developed [Figure 5] and are under investigation for prostate cancer include:
Suicide gene therapy
In this method, a gene encoding an enzyme that can change a prodrug from harmless to toxic is delivered by a viral vector. When the prodrug is given systemically, it kills tumor cells that produce the enzyme specifically, leaving healthy cells unharmed. Thymidine kinase (HSV-tk), for instance, is an enzyme that may phosphorylate ganciclovir, a nucleoside analog that stops DNA synthesis and causes apoptosis in herpes simplex virus infection. Patients with prostate cancer localized to a particular locality, who participated in a clinical experiment wherein HSV-tk was delivered into the prostate gland via an adenovirus vector, experienced a noteworthy decrease in both tumor volume and PSA levels[269,302].
Tumor suppressor gene therapy
This method delivers a gene that encodes a tumor suppressor protein, a protein that can control apoptosis, the cell cycle, or DNA repair, using a viral vector. When endogenous tumor suppressor genes are deleted or altered in tumor cells, the tumor suppressor protein can either induce cell death or restore normal cell function. A tumor suppressor protein called REIC/Dkk-3 has the ability to block Wnt signaling and trigger endoplasmic reticulum stress-mediated apoptosis. In a clinical experiment, patients suffering from castration-resistant prostate cancer experienced notable reductions in tumor volume and PSA levels following the delivery of REIC/Dkk-3 to their prostate glands using an adenovirus vector[303].
Cytokine gene therapy
This method delivers a gene that codes for a cytokine, a protein that can influence the immune system and have antitumor properties, via a viral vector. The cytokine can suppress tumor development and angiogenesis, improve antigen presentation, and activate immune cells both locally and systemically. In a rat model of prostate cancer, employing an adenovirus vector to deliver the cytokine interleukin-12 to TAMs could cause their reprogramming from an M2-like to an M1-like phenotype and prevent tumor growth and metastasis[304].
Viral vector-gene therapy offers the potential for straightforward tumor destruction without the requirement for long-lasting transgene expression, serving as a targeted approach for various types of prostate cancer. Apart from intratumoral delivery, experiments have been conducted on tumor targeting using vectors particularly engineered for that purpose, which involves using tumor-specific promoters and utilizing oncolytic viruses[288].
Corrective gene therapy
One kind of gene treatment called "corrective gene therapy" tries to fix the genetic flaws or mutations that either cause or aggravate prostate cancer. It operates under the premise that cancer cells can be stopped from proliferating and spreading by injecting a functional or normal copy of a gene into them[294]. Viral or nonviral transport vehicles are used to transfer the genes to the target cells in corrective gene therapy.
Using adenovirus vectors to transmit the p53 gene - a tumor suppressor gene frequently altered or destroyed in prostate cancer - is one instance of corrective gene therapy for the disease[294,303].
Senescence, apoptosis, or cell cycle arrest can all be brought on by the p53 gene in reaction to stress signals or DNA damage. Corrective gene therapy can cause prostate cancer cells to self-destruct and stop growing by restoring the expression of p53 in such cells.
The delivery of the PTEN gene, another tumor suppressor gene frequently lost or deactivated in prostate cancer, using adeno-associated virus (AAV) vectors is another example of corrective gene therapy for the disease. Through antagonistic action on the PI3K/AKT signaling pathway, frequently hyperactivated in prostate cancer, the PTEN gene can control cell proliferation, survival, and invasion. Corrective gene therapy can lessen the multiplication and invasiveness of prostate cancer cells by restoring the response of PTEN in those cells[294,305].
Prostate cancer corrective gene therapy is currently in the experimental stage and has not yet received clinical approval. There are ongoing or completed clinical trials calculating the effectiveness and safety of this methodology. For instance, in a trial of gene-directed enzyme prodrug therapy (GDEPT) for early prostate cancer, a gene that can activate the innocuous medication CB1954 into a potent anti-cancer drug was delivered by a specially treated virus. Additionally, for locally advanced prostate cancer, the conjunction of adenovirus-mediated p53 gene therapy with radiation therapy was investigated in different trials[305]. Although these trials have yielded some encouraging findings, further investigation is required to address the obstacles and restrictions associated with corrective gene therapy, including limited transfection efficiency, immunological response, specificity, and toxicity[294,305].
Potent nonviral gene therapies in prostate cancer
Since multiple genes in malignant cells are impacted by different genetic abnormalities, cancer is thought to be a genetic condition. Tumor suppressor genes such as TP53 and NM23, and oncogenes such as RAS, c-MYC, BCL2, and c-MET are the most commonly affected. Tumor suppressor genes play a crucial role in regulating normal cell death and the elimination of cellular waste products. Deactivation of these genes, through mutation or absence, can promote cell malignancy. Oncogenes, on the other hand, are responsible for maintaining steady cell proliferation and their activation can promote cancer cell growth[293]. Although genome editing medicines have traditionally been delivered using viral vectors in lab settings and clinical trials, their translation has been severely hampered by the possible immunogenic risks associated with viral carriers. However, nonviral administration methods have emerged as a promising alternative. Nonviral delivery systems, intentionally synthesized and with fewer safety concerns, offer a viable option. Although they have somewhat lower delivery efficiency compared to viral delivery systems, they present a safer alternative with reduced immunogenic risks.
Numerous genes are implicated in genetic alterations in prostate cancer. For example, it was discovered that approximately fifty and thirty-five percent of advanced-stage prostate cancer cases exhibit mutations in the tumor suppressor gene TP53 and retinoblastoma, respectively[293].
Mutations in genes cause uncontrollable cell development. TP53 primarily plays a role in maintaining the cell life cycle and repairing DNA damage. Additionally, it has been discovered that certain cases of prostate cancer had disruptions in the GSTP1 gene. GSTP1's primary physiological function is carcinogen detoxification, and its deactivation promotes carcinogenesis[293]. According to applications, potent nonviral gene therapies include:
Direct tumor injection
Direct tumor injection aims to eradicate prostate cancer cells by delivering genetic material directly into tumors. In a phase-I trial, the efficacy of an IL-2 plasmid with a cytomegalovirus (CMV) promoter bound to lipid-bilayer carrier molecules was evaluated in individuals with high-risk prostate cancer[306].
Qualified patients either experienced cancer recurrence after upfront radiation therapy or cryotherapy, or had advanced localized disease (confirmed by Gleason score 7 or higher and PSA greater than 10 ng mL-1). On days 1 and 7 of the research, transrectal intra-prostatic gene delivery was performed by injecting the lipid bilayer molecules complexed with DNA into hypoechoic lesions identified by transrectal ultrasonography (TRUS) in all males with biopsy-proven lesions. Of the twenty-four patients receiving treatment, ten had recurrent illnesses and fourteen had localized illnesses. The therapy was well tolerated, with 16 patients experiencing a brief drop in PSA levels on the first day after treatment. Furthermore, there was evidence of enhanced peripheral blood lymphocyte proliferation and local T cell infiltration post-injection. The expression of IL-2 DNA and mRNA persisted up to seven days after injection, likely due to the transient effect of plasmid transporter vehicles in situ. This indicates that the plasmid system had a temporary impact[306,307]. These data are promising, as some studies suggest that this therapy is a more favorable therapy for the treatment of early-stage prostate cancer.
Gene apoptosis therapy
Prostate cancer stands as a leading cause of mortality among men worldwide. Current treatment options such as radiation, hormone therapy, and prostatectomy often come with unfavorable side effects and varying levels of effectiveness across different stages of the disease. In this context, gene therapy emerges as a promising and experimental approach for treating prostate cancer. One notable strategy within gene therapy involves inducing cellular apoptosis in cancer cells[293,308,309]. A significant challenge posed by genetic alterations in cancer cells is commonly the suppression of apoptosis, contributing to cancer progression[309]. However, the lack of delivery technologies that can selectively deliver therapeutic genes to tumors without causing significant adverse effects following intravenous treatment currently limits its application[293]. To address this limitation, numerous nonviral delivery strategies have been developed. These strategies aim to deliver DNA-based medicinal medicines precisely to the intended sites of action[293].
Immunomodulatory gene therapy
Immunomodulatory gene therapy is a type of gene treatment aimed at enhancing the immune system’s ability to detect and destroy prostate cancer cells. The premise underlying this approach is the activation of the antitumor response of the immune system and the subversion of the immunosuppressive mechanisms of cancer cells by introducing genes into the TME that stimulate the production of immune mediators such as cytokines, chemokines, or costimulatory molecules[311]. Both viral and nonviral vehicles are used as carriers to deliver therapeutic genes to target cancer cells in immunomodulatory gene therapy[294,310].
An example of immunomodulatory gene therapy for prostate cancer involves the use of plasmid DNA vectors to transport the interleukin-12 (IL-12) gene. IL-12 induces the production of a potent pro-inflammatory cytokine, activating the innate and adaptive immune systems to combat tumor growth.
The balance between pro- and antitumor cytokines can also be influenced by IL-12 production in the TME, which promotes the infiltration and activation of NK (natural killer) cells, macrophages, dendritic cells, and T cells[294,311]. For prostate cancer, a phase I trial combining this treatment with radiation therapy has demonstrated its viability, tolerability, and early signs of antitumor activity[311].
Despite promising results, immunomodulatory gene therapy for prostate cancer has not yet received clinical approval. Various clinical trials are underway evaluate the effectiveness and safety in combination with other treatments such as radiation, chemotherapy, hormone therapy, and immunotherapy. These trials seek to optimize delivery strategies, dosing regimens, gene combinations, and treatment sequences, as well as to identify potential biomarkers of resistance and response[294,311].
RECENT PROGRESS IN GENE THERAPY FOR PROSTATE CANCER
Recent developments in gene therapy have shown promising results in the fight against prostate cancer, particularly through the authorization of rucaparib and Olaparib. These drugs, known as PARP (poly ADP-ribose polymerase) inhibitors, are effective in subjects with mutations in DNA damage repair genes, offering new hope for individuals with metastatic Castration-Resistant Prostate Cancer (mCRPC). By blocking the enzyme responsible for DNA repair in cancer cells, PARP inhibitors induce cell death. Specifically, olaparib has been found to delay disease progression, alleviate pain, and potentially extend the lives of patients with mutations in the BRCA1, BRCA2, and ATM genes, as evidenced by a large, multinational phase 3 trial[312]. Furthermore, rucaparib has shown comparable benefits for subjects with mCRPC harboring mutations in genes such as BRCA1, BRCA2, or PALB2[313].
Beyond these therapies, pembrolizumab, an immunotherapy medication that stimulates T cells to combat cancer cells, and 177-lutetium-PSMA-617, a radiotherapeutic agent that targets PSMA-positive prostate cancer cells, are currently undergoing clinical trials. These trials aim to explore the potential of nonviral gene transfer methods for treating prostate cancer cells in patients with mCRPC who have responded to traditional therapy[295]. Nonviral transfer vehicles, while presenting certain drawbacks, such as low transfection efficiency, poor specificity, and limited duration of gene expression, are generally considered less immunogenic, less toxic, and more adaptable than viral vectors. Recent advancements in this area include the use of lipid nanoparticles (LNPs) for delivering mRNA encoding various proteins, polymeric micelles for CRISPR/Cas9-mediated gene editing, and bisphosphonate nanocomplexes for siRNA targeting different genes[284,305-307].
One notable advancement involves the use of RALA peptide as a nonviral vector to deliver plasmids expressing inducible nitric oxide synthase (iNOS), a powerful antitumor agent that promotes cancer cell apoptosis and inhibits angiogenesis in prostate cancer cells. A study has demonstrated that RALA/iNOS composite nanoparticles are both safer and more effective in combating tumors compared to RALA alone or free iNOS plasmids[314].
DISCUSSION
Over the past few decades, tremendous progress has been made in prostate cancer treatment. However, navigating the complexities of employing immune therapy in prostate cancer presents both challenges and potential breakthroughs. Despite the initial attractiveness of targeting the disease due to its slow progression, clinical trials, especially in metastatic castration-resistant prostate cancer (mCRPC), face hurdles in achieving optimal efficacy. These challenges stem from compromised immune systems in patients, characterized by deficiencies in cellular immunity and reduced natural killer cell activity. Additionally, the intricate microenvironment of prostate tumors further complicates immune cell function. Nevertheless, emerging evidence and advancements in CAR-T therapy offer hope for tackling challenging cases. A comprehensive understanding of cytokines, chemokines, and immune cell dynamics is imperative for developing effective immunotherapies[315,316]. Despite acknowledged limitations, cautious optimism prevails regarding the future of immunotherapy in advanced prostate cancer.
Prostate cancer treatment encounters substantial hurdles due to its complex interactions with the immune system. The tumor microenvironment fosters myeloid-derived suppressor cells (MDSCs), T regulatory cells (Tregs), and tumor-associated macrophages (TAMs), hindering effector T cell functions. Grasping these evasion mechanisms is crucial. The TASQ trial's focus on targeting MDSCs exhibits potential in cancer immunotherapy. Tumors strategically create an immune-tolerant microenvironment, impeding immune-based therapies, countered by approaches like combining vaccines with agents such as imatinib and sunitinib. The diverse roles of T-regulatory cells (Tregs) and Th17 cells suggest promising avenues for enhancing treatment effectiveness. Striking a balance in these dynamics could drive progress in prostate cancer immunotherapy, necessitating further research and clinical validation.
Additionally, the complexities of treating prostate cancer have led to the exploration of diverse immunotherapy targets. Key proteins such as PSA, widely used as a serum marker, show promise by triggering the activation of tumor-reactive CD8+ cytotoxic T cells. PSMA, highly expressed in prostate tumors, attains significance as both a marker and a therapeutic target, securing FDA approval for advanced metastatic prostate cancer. PAP, plentiful in prostate tissue, displays potential in immunotherapy, particularly evident in sipuleucel-T trials. Other targets such as PSCA, dMMR, and prostein have demonstrated positive outcomes in both preclinical and clinical settings. A comprehensive approach involving Trp-p8, STEAP1, and NY-ESO-1 highlights the evolving landscape of prostate cancer immunotherapy, emphasizing the ongoing importance of research and clinical validation. Overexpressed proteins in prostate cancer, including PTHrP, hTERT, survivin, and members of the EGFR family, present viable targets for immunotherapy[317]. Promising interventions, such as monoclonal antibodies, dendritic cell vaccines, and inhibitors, demonstrate potential in preclinical models. EphA2 and SSX are under investigation for active immunotherapy, while EpCAM and RIPK2 remain potential targets with acknowledged challenges. Continued research is essential to optimize approaches and improve patient outcomes in prostate cancer treatment.
In-depth studies on immune dynamics in advanced prostate cancer reveal its dual role, acting as both a defense against intruders and inadvertently supporting tumor growth. The classification of cancers into hot or cold tumors depends on immune infiltration. Prostate cancer, identified as a cold tumor due to its low immune density, encounters obstacles such as MDSCs and TME suppression. The significant impact of bones on tumor development and immunotherapy underscores the necessity for precision in treatment strategies. Effectively addressing immune-suppressive challenges, notably the prevalence of MDSCs, is imperative for enhancing treatment outcomes.
Revolutionary immune checkpoint inhibitors (ICIs) fortify the immune system by obstructing proteins on immune or cancer cells, facilitating the identification and eradication of cancer cells. Prostate cancer presents unique challenges due to factors such as low mutation rates, restricted PD-L1 expression, and intensified immunosuppression, which can complicate the efficacy of ICIs. Although specific prostate cancer subtypes show promise, uncertainties persist, underscoring the need for ongoing research to refine treatment approaches. The FDA approval of Sipuleucel-T for metastatic castrate-resistant prostate cancer signifies progress, emphasizing further exploration into biomarkers, efficacy, and toxicity to improve patient outcomes.
Advanced prostate cancer poses substantial challenges to immunotherapy. The obstacles include sparse neoantigens hindering immune response, low antigen levels complicating recognition, and an immunosuppressive microenvironment. Tumor heterogeneity, DNA repair issues, and dependence on the androgen receptor further complicate existing immunotherapies. Resistance to checkpoint inhibitors, involving macrophage-driven IL-23 and STAT3 activation, underscores the demand for precision approaches. Continuous research strives to optimize immunotherapy outcomes in prostate cancer.
Moreover, gene editing techniques utilizing nucleases such as base editors, ZFNs, CRISPR/Cas9, and TALENs enable precise modifications to genes associated with prostate cancer. Enhanced therapeutic efficacy is achieved through diverse gene delivery methods, encompassing both viral and nonviral vectors [Table 2]. Innovative strategies such as suicide gene therapy and immunomodulation contribute to the dynamic evolution of prostate cancer treatment, offering considerable potential for advanced therapeutic outcomes.
Targeting prostate cancer using viral vector-based gene therapy involves the utilization of various viral vectors such as measles virus (MV), mumps virus, alphavirus, Venezuelan equine encephalitis virus (VEEV), and Vaccinia virus (VV GLV-1h123), as well as the viral replicon vector system (alphavirus, Venezuelan equine encephalitis virus). These vectors facilitate the delivery of therapeutic molecules carrying different target genes into subjects (prostate cancer patients, mice)
| Vector | Subject/model organism | Therapeutic molecules | Target gene | Inference |
| Measles virus (MV) | Mice | Measles virus strains deriving from the Edmonston (MV-Edm) vaccine strain | Carcinoembryonic antigen (CEA) | Intraperitoneal injection of MV-CEA particles inhibited tumor formation and prolonged the lifespan of mice with prostate tumor PC-3, a condition associated with prostate cancer[318] |
| Measles virus (MV) | Mice | Single-chain antibody (sc-Fv) | Prostate-specific membrane antigen (PSMA) | In a distinct study, mice bearing PC3-PSMA and LNCaP prostate cancers were administered a measles virus (MV vector) containing a specific sc-Fv focusing on the outer domain of the prostate-specific membrane antigen[319] |
| MV, mumps virus (MuV) | Mice | single-chain antibody (sc-Fv) | Prostate-specific membrane antigen (PSMA) | Radiation therapy improved the targeted disease and breaking of PSMA-positive prostate cancer cells that MV-sc-Fv-PSMA delivered. When oncolytic MV and MuV vehicles were administered together, PC-3 in mice prostate cancers exhibited increased anticancer activity and longer survival times compared to when MV or MuV vectors were administered separately[320] |
| Alphavirus | BALB/c C57BL/6 mice | Venezuelan equine encephalitis (VEE) particle | Prostate-specific membrane antigen (PSMA) | Another method involved the PSMA in BALB/c and C57BL/6 mice cells using VEE, which resulted in robust immune activity specific to PSMA[169]. Strong, Th1-biased T and B cell responses were induced by a single vaccine |
| Venezuelan equine encephalitis virus (VEEV) | Mice | Venezuelan equine encephalitis (VEE) particle | Mouse-six-Trans-membrane Epithelial Antigen of the Prostate (mSTEAP). | Following an initial immunization using conventional pcDNA-3-mSTEAP plasmids coated with gold, mice were given increment immunization using VEE particles that showed mouse mSTEAP. This approach triggered specific immune responses against mSTEAP, resulting in a slight yet noteworthy slowdown in tumor growth and extending the overall survival of the mice[124] |
| Venezuelan equine encephalitis virus (VEEV) | Transgenic mice | Venezuelan equine encephalitis (VEE) particle | Prostate stem cell antigen (PSCA) | 90% of transgenic mice with prostate cancer (TRAMP) exhibited long-term survival after receiving VEE molecules with PSCA[321] |
| Vaccinia virus (VV GLV-1h123) | Mice | NIS (Sodium iodide symporter) protein | Gene: Sodium iodide symporter (NIS) | In male mice models of prostate cancer, delivery of VV GLV-1h123 vehicle, which expresses the sodium iodide symporter (NIS) gene, resulted in a significant reduction in tumor growth and an extension of lifespan[322] |
| Viral replicon vector system (alphavirus, Venezuelan equine encephalitis virus) | Subjects bearing CRPC | Venezuelan equine encephalitis (VEE) particle | PSMA | Phase I trial using VEE-PSMA particles was followed in subjects with CRPC for clinical evaluation[323] |
CONCLUSION
Despite strides in immunotherapy and gene therapy, overcoming the immunosuppressive traits of tumors and ensuring consistent treatment responses remains challenging. The necessity for tailored and integrated approaches is underscored, offering hope for future strategies aimed at improving therapeutic outcomes for prostate cancer patients. Comprehending the array of overexpressed and predominantly expressed proteins in prostate cancer provides potential avenues for immunotherapeutic interventions. Each protein presents distinct challenges and prospects, underscoring the imperative for ongoing research to refine treatment modalities and enhance patient outcomes. The integration of precise gene editing techniques with efficient gene delivery methods holds great promise for advancing prostate cancer treatment. Targeted strategies and continuous research are essential to tackle challenges and optimize outcomes in the evolving landscape of prostate cancer gene therapy. Examining combination therapy strategies, particularly the integration of immunotherapy with chemotherapy, radiation, or hormone therapy, holds potential for identifying synergistic effects. Such investigations may open new avenues to enhance treatment efficacy in prostate cancer. The exploration of potential new targets, creative delivery approaches, and innovative therapy combinations is paramount. By highlighting these future prospects, it will guide research efforts and spark fresh ideas in the field.
DECLARATIONS
Authors’ contributions
Conceived and designed the study; performed data collection and analysis; drafted the manuscript; revised the manuscript critically for important intellectual content; approved the final version to be published:
Contributed to the conceptualization of the study; contributed to data collection and analysis; Reviewed and provided feedback on the manuscript; approved the final version to be published: Sarkar K
Both authors contributed to writing and critically revising the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024
REFERENCES
1. Rehman LU, Nisar MH, Fatima W, et al. Immunotherapy for prostate cancer: a current systematic review and patient centric perspectives. J Clin Med 2023;12:1446.
2. Tewari AK, Stockert JA, Yadav SS, Yadav KK, Khan I. Inflammation and prostate cancer. Adv Exp Med Biol 2018;1095:41-65.
3. Gurel B, Lucia MS, Thompson IM Jr, et al. Chronic inflammation in benign prostate tissue is associated with high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol Biomarkers Prev 2014;23:847-56.
4. Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology 2012;60:199-215.
5. Sfanos KS, Isaacs WB, De Marzo AM. Infections and inflammation in prostate cancer. Am J Clin Exp Urol 2013;1:3-11.
6. Cai T, Santi R, Tamanini I, et al. Current knowledge of the potential links between inflammation and prostate cancer. Int J Mol Sci 2019;20:3833.
7. Saxby H, Boussios S, Mikropoulos C. Androgen receptor gene pathway upregulation and radiation resistance in oligometastatic prostate cancer. Int J Mol Sci 2022;23:4786.
8. Sfanos KS, Bruno TC, Maris CH, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res 2008;14:3254-61.
9. Bubendorf L, Schöpfer A, Wagner U, et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol 2000;31:578-83.
10. Bilusic M, Madan RA, Gulley JL. Immunotherapy of prostate cancer: facts and hopes. Clin Cancer Res 2017;23:6764-70.
11. Markowski MC, Shenderov E, Eisenberger MA, et al. Extreme responses to immune checkpoint blockade following bipolar androgen therapy and enzalutamide in patients with metastatic castration resistant prostate cancer. Prostate 2020;80:407-11.
12. Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803-13.
13. Luke JJ, Rutkowski P, Queirolo P, et al. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): a randomised, double-blind, phase 3 trial. Lancet 2022;399:1718-29.
14. Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 2018;378:1277-90.
15. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23-34.
16. Petrylak DP, de Wit R, Chi KN, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel in patients with locally advanced or metastatic urothelial carcinoma after platinum-based therapy (RANGE): a randomised, double-blind, phase 3 trial. Lancet 2017;390:2266-77.
17. Lee NY, Ferris RL, Psyrri A, et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol 2021;22:450-62.
18. Boussios S, Rassy E, Shah S, Ioannidou E, Sheriff M, Pavlidis N. Aberrations of DNA repair pathways in prostate cancer: a cornerstone of precision oncology. Expert Opin Ther Targets 2021;25:329-33.
19. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003;302:415-9.
22. Catalona WJ, Smith DS, Ratliff TL, et al. Measurement of prostate-specific antigen in serum as a screening test for prostate cancer. N Engl J Med 1991;324:1156-61.
23. Cooperberg MR, Broering JM, Carroll PR. Risk assessment for prostate cancer metastasis and mortality at the time of diagnosis. J Natl Cancer Inst 2009;101:878-87.
24. Koeneman KS, Kao C, Ko SC, et al. Osteocalcin-directed gene therapy for prostate-cancer bone metastasis. World J Urol 2000;18:102-10.
25. Madan RA, Gulley JL. Finding an immunologic beachhead in the prostate cancer microenvironment. J Natl Cancer Inst 2019;111:219-20.
26. Liu G, Lu S, Wang X, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest 2013;123:4410-22.
27. Wu JD, Higgins LM, Steinle A, Cosman D, Haugk K, Plymate SR. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest 2004;114:560-8.
28. Healy CG, Simons JW, Carducci MA, et al. Impaired expression and function of signal-transducing zeta chains in peripheral T cells and natural killer cells in patients with prostate cancer. Cytometry 1998;32:109-19.
29. Pal SK, Moreira D, Won H, et al. Reduced T-cell numbers and elevated levels of immunomodulatory cytokines in metastatic prostate cancer patients de novo resistant to abiraterone and/or enzalutamide therapy. Int J Mol Sci 2019;20:1831.
30. Lopez-Bujanda Z, Drake CG. Myeloid-derived cells in prostate cancer progression: phenotype and prospective therapies. J Leukoc Biol 2017;102:393-406.
31. Idorn M, Køllgaard T, Kongsted P, Sengeløv L, Thor Straten P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol Immunother 2014;63:1177-87.
32. Chi N, Tan Z, Ma K, Bao L, Yun Z. Increased circulating myeloid-derived suppressor cells correlate with cancer stages, interleukin-8 and -6 in prostate cancer. Int J Clin Exp Med 2014;7:3181-92.
33. Calagua C, Russo J, Sun Y, et al. Expression of PD-L1 in hormone-naïve and Treated prostate cancer patients receiving neoadjuvant abiraterone acetate plus prednisone and leuprolide. Clin Cancer Res 2017;23:6812-22.
34. Shariat SF, Kim JH, Andrews B, et al. Preoperative plasma levels of transforming growth factor β1 strongly predict clinical outcome in patients with bladder carcinoma. Cancer 2001;92:2985-92.
35. Miller AM, Lundberg K, Ozenci V, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol 2006;177:7398-405.
36. Yokokawa J, Cereda V, Remondo C, et al. Enhanced functionality of CD4+CD25highFoxP3+ regulatory T cells in the peripheral blood of patients with prostate cancer. Clin Cancer Res 2008;14:1032-40.
37. Getnet D, Maris CH, Hipkiss EL, et al. Tumor recognition and self-recognition induce distinct transcriptional profiles in antigen-specific CD4 T cells. J Immunol 2009;182:4675-85.
38. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009;182:4499-506.
39. Zlotta AR, Egawa S, Pushkar D, et al. Prevalence of prostate cancer on autopsy: cross-sectional study on unscreened Caucasian and Asian men. J Natl Cancer Inst 2013;105:1050-8.
40. Whittemore AS, Cirillo PM, Feldman D, Cohn BA. Prostate specific antigen levels in young adulthood predict prostate cancer risk: results from a cohort of Black and White Americans. J Urol 2005;174:872-6; discussion 876.
41. Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer 2018;6:157.
42. Perera MPJ, Thomas PB, Risbridger GP, et al. Chimeric antigen receptor T-Cell therapy in metastatic castrate-resistant prostate cancer. Cancers 2022;14:503.
43. Mughees M, Kaushal JB, Sharma G, Wajid S, Batra SK, Siddiqui JA. Chemokines and cytokines: axis and allies in prostate cancer pathogenesis. Semin Cancer Biol 2022;86:497-512.
44. Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol 2006;90:51-81.
45. Tien AH, Xu L, Helgason CD. Altered immunity accompanies disease progression in a mouse model of prostate dysplasia. Cancer Res 2005;65:2947-55.
47. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9:162-74.
48. Mortezaee K. Myeloid-derived suppressor cells in cancer immunotherapy-clinical perspectives. Life Sci 2021;277:119627.
49. Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res 2010;70:5679-85.
50. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002;3:651-62.
51. Vuk-Pavlović S, Bulur PA, Lin Y, et al. Immunosuppressive CD14+HLA-DRlow/- monocytes in prostate cancer. Prostate 2010;70:443-55.
52. Gustafson MP, Lin Y, League SC, et al. Loss of HLA-DR expression on CD14+cells; A common marker of immunosuppression in cancer patients. J Immunother 2010;33:869-70. Available from: https://www.researchgate.net/publication/295679244_Loss_of_HLA-DR_Expression_on_CD14Cells_A_Common_Marker_of_Immunosuppression_in_Cancer_Patients [Last accessed on 12 Apr 2024]
53. Pili R, Häggman M, Stadler WM, et al. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol 2011;29:4022-8.
54. Björk P, Björk A, Vogl T, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol 2009;7:e97.
55. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008;8:618-31.
56. Thakur A, Vaishampayan U, Lum LG. Immunotherapy and immune evasion in prostate cancer. Cancers 2013;5:569-90.
57. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 2007;25:267-96.
58. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 2012;12:237-51.
59. Balachandran VP, Cavnar MJ, Zeng S, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 2011;17:1094-100.
60. Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 2009;69:2514-22.
61. Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 2004;34:336-44.
62. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 2005;11:6713-21.
63. Hurwitz AA, Foster BA, Kwon ED, et al. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res 2000;60:2444-8.
64. Hiura T, Kagamu H, Miura S, et al. Both regulatory T cells and antitumor effector T cells are primed in the same draining lymph nodes during tumor progression. J Immunol 2005;175:5058-66.
65. Tang S, Moore ML, Grayson JM, Dubey P. Increased CD8+ T-cell function following castration and immunization is countered by parallel expansion of regulatory T cells. Cancer Res 2012;72:1975-85.
66. Vergati M, Cereda V, Madan RA, et al. Analysis of circulating regulatory T cells in patients with metastatic prostate cancer pre- versus post-vaccination. Cancer Immunol Immunother 2011;60:197-206.
67. Derhovanessian E, Adams V, Hähnel K, et al. Pretreatment frequency of circulating IL-17+ CD4+ T-cells, but not Tregs, correlates with clinical response to whole-cell vaccination in prostate cancer patients. Int J Cancer 2009;125:1372-9.
68. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009;9:353-63.
69. Lundwall A, Lilja H. Molecular cloning of human prostate specific antigen cDNA. FEBS Lett 1987;214:317-22.
70. Ruan H, Bao L, Tao Z, Chen K. Flightless I homolog reverses enzalutamide resistance through PD-L1-mediated immune evasion in prostate cancer. Cancer Immunol Res 2021;9:838-52.
71. Oesterling JE. Prostate specific antigen: a critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J Urol 1991;145:907-23.
73. Freedland SJ, Hotaling JM, Fitzsimons NJ, et al. PSA in the new millennium: a powerful predictor of prostate cancer prognosis and radical prostatectomy outcomes-results from the SEARCH database. Eur Urol 2008;53:758-64; discussion 765.
74. Xue B, Zhang Y, Sosman JA, Peace DJ. Induction of human cytotoxic T lymphocytes specific for prostate-specific antigen. Prostate 1997;30:73-8.
75. Correale P, Nieroda C, Zaremba S, et al. In vitro generation of human cytotoxic T lymphocytes specific for peptides derived from prostate-specific antigen. J Natl Cancer Inst 1997;89:293-300.
76. Perambakam S, Xue BH, Sosman JA, Peace DJ. Induction of Tc2 cells with specificity for prostate-specific antigen from patients with hormone-refractory prostate cancer. Cancer Immunol Immunother 2002;51:263-70.
77. Terasawa H, Tsang KY, Gulley J, Arlen P, Schlom J. Identification and characterization of a human agonist cytotoxic T-lymphocyte epitope of human prostate-specific antigen. Clin Cancer Res 2002;8:41-53.
78. Heiser A, Dahm P, Yancey DR, et al. Human dendritic cells transfected with RNA encoding prostate-specific antigen stimulate prostate-specific CTL responses in vitro. J Immunol 2000;164:5508-14.
79. Correale P, Walmsley K, Zaremba S, Zhu M, Schlom J, Tsang KY. Generation of human cytolytic T lymphocyte lines directed against prostate-specific antigen (PSA) employing a PSA oligoepitope peptide. J Immunol 1998;161:3186-94.
80. Harada M, Kobayashi K, Matsueda S, Nakagawa M, Noguchi M, Itoh K. Prostate-specific antigen-derived epitopes capable of inducing cellular and humoral responses in HLA-A24+ prostate cancer patients. Prostate 2003;57:152-9.
81. Gotoh M, Takasu H, Harada K, Yamaoka T. Development of HLA-A2402/Kb transgenic mice. Int J Cancer 2002;100:565-70.
82. Corman JM, Sercarz EE, Nanda NK. Recognition of prostate-specific antigenic peptide determinants by human CD4 and CD8 T cells. Clin Exp Immunol 1998;114:166-72.
83. Matsueda S, Takedatsu H, Yao A, et al. Identification of peptide vaccine candidates for prostate cancer patients with HLA-A3 supertype alleles. Clin Cancer Res 2005;11:6933-43.
84. Klyushnenkova EN, Link J, Oberle WT, et al. Identification of HLA-DRB1*1501-restricted T-cell epitopes from prostate-specific antigen. Clin Cancer Res 2005;11:2853-61.
85. Mahadevan M, Liu Y, You C, et al. Generation of robust cytotoxic T lymphocytes against prostate specific antigen by transduction of dendritic cells using protein and recombinant adeno-associated virus. Cancer Immunol Immunother 2007;56:1615-24.
86. Arredouani MS, Tseng-Rogenski SS, Hollenbeck BK, et al. Androgen ablation augments human HLA2.1-restricted T cell responses to PSA self-antigen in transgenic mice. Prostate 2010;70:1002-11.
87. Drake CG, Doody AD, Mihalyo MA, et al. Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell 2005;7:239-49.
88. Cornford P, van den Bergh RCN, Briers E, et al. Eau-Eanm-Estro-Esur-Siog guidelines on prostate cancer. Part II-2020 update: treatment of relapsing and metastatic prostate cancer. Eur Urol 2021;79:263-82.
89. Just J, Osgun F, Knight C. Lower urinary tract symptoms and prostate cancer: is PSA testing in men with symptoms wise? Br J Gen Pract 2018;68:541-2.
90. Troyer JK, Beckett ML, Wright GL Jr. Detection and characterization of the prostate-specific membrane antigen (PSMA) in tissue extracts and body fluids. Int J Cancer 1995;62:552-8.
91. Israeli RS, Powell CT, Corr JG, Fair WR, Heston WD. Expression of the prostate specific membrane antigen. Cancer Res 1994;54:1807-11.
92. Sokoloff RL, Norton KC, Gasior CL, Marker KM, Grauer LS. A dual-monoclonal sandwich assay for prostate-specific membrane antigen: levels in tissues, seminal fluid and urine. Prostate 2000;43:150-7.
93. Murphy GP, Elgamal AA, Su SL, Bostwick DG, Holmes EH. Current evaluation of the tissue localization and diagnostic utility of prostate specific membrane antigen. Cancer 1998;83:2259-69.
94. Kawakami M, Nakayama J. Enhanced expression of prostate-specific membrane antigen gene in prostate cancer as revealed by in situ hybridization. Cancer Res 1997;57:2321-4.
95. Kuang Y, Weng X, Liu X, et al. Anti-tumor immune response induced by dendritic cells transduced with truncated PSMA IRES 4-1BBL recombinant adenoviruses. Cancer Lett 2010;293:254-62.
96. George DJ. An α-particle emitting antibody ([213Bi]J591) for radioimmunotherapy of prostate cancer. Prostate 2001;3:1.
97. Fracasso G, Bellisola G, Cingarlini S, et al. Anti-tumor effects of toxins targeted to the prostate specific membrane antigen. Prostate 2002;53:9-23.
98. Vallabhajosula S, Smith-Jones PM, Navarro V, Goldsmith SJ, Bander NH. Radioimmunotherapy of prostate cancer in human xenografts using monoclonal antibodies specific to prostate specific membrane antigen (PSMA): studies in nude mice. Prostate 2004;58:145-55.
99. Carroll PR. Radiolabeled monoclonal antibodies specific to the extracellular domain of prostate-specific membrane antigen: preclinical studies in nude mice bearing LNCaP human prostate tumor: Smith-Jones PM, Vallabhajosula S, Navarro V, Bastidas D, Goldsmith SJ, Bander NH, Division of nuclear medicine, department of radiology, New York presbyterian Hospital-Weill Medical College of Cornell University, New York, NY. J Nucl Med 2003;44:610-617. Urol Oncol-Semin Origin Invest 2003;21:486-7.
100. Kuroda K, Liu H, Kim S, Guo M, Navarro V, Bander NH. Saporin toxin-conjugated monoclonal antibody targeting prostate-specific membrane antigen has potent anticancer activity. Prostate 2010;70:1286-94.
101. Sartor O, Baghian A. Prostate specific membrane antigen binding radiopharmaceuticals: current data and new concepts. Front Med 2022;9:1060922.
102. Solin T, Kontturi M, Pohlmann R, Vihko P. Gene expression and prostate specificity of human prostatic acid phosphatase (PAP): evaluation by RNA blot analyses. Biochim Biophys Acta 1990;1048:72-7.
103. Cunha AC, Weigle B, Kiessling A, Bachmann M, Rieber EP. Tissue-specificity of prostate specific antigens: comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett 2006;236:229-38.
104. Graddis TJ, McMahan CJ, Tamman J, Page KJ, Trager JB. Prostatic acid phosphatase expression in human tissues. Int J Clin Exp Pathol 2011;4:295-306.
105. Machlenkin A, Paz A, Bar Haim E, et al. Human CTL epitopes prostatic acid phosphatase-3 and six-transmembrane epithelial antigen of prostate-3 as candidates for prostate cancer immunotherapy. Cancer Res 2005;65:6435-42.
106. Machlenkin A, Azriel-Rosenfeld R, Volovitz I, et al. Preventive and therapeutic vaccination with PAP-3, a novel human prostate cancer peptide, inhibits carcinoma development in HLA transgenic mice. Cancer Immunol Immunother 2007;56:217-26.
107. Olson BM, Frye TP, Johnson LE, et al. HLA-A2-restricted T-cell epitopes specific for prostatic acid phosphatase. Cancer Immunol Immunother 2010;59:943-53.
108. Spies E, Reichardt W, Alvarez G, Groettrup M, Ohlschläger P. An artificial PAP gene breaks self-tolerance and promotes tumor regression in the TRAMP model for prostate carcinoma. Mol Ther 2012;20:555-64.
109. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411-22.
110. Kiessling A, Wehner R, Füssel S, Bachmann M, Wirth MP, Schmitz M. Tumor-associated antigens for specific immunotherapy of prostate cancer. Cancers 2012;4:193-217.
111. Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502-12.
112. Schellhammer PF, Chodak G, Whitmore JB, Sims R, Frohlich MW, Kantoff PW. Lower baseline prostate-specific antigen is associated with a greater overall survival benefit from sipuleucel-T in the Immunotherapy for Prostate Adenocarcinoma Treatment (IMPACT) trial. Urology 2013;81:1297-302.
113. Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367:1187-97.
114. Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004;351:1513-20.
115. Sheikh NA, Petrylak D, Kantoff PW, et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol Immunother 2013;62:137-47.
116. McNeel DG, Becker JT, Eickhoff JC, et al. Real-time immune monitoring to guide plasmid DNA vaccination schedule targeting prostatic acid phosphatase in patients with castration-resistant prostate cancer. Clin Cancer Res 2014;20:3692-704.
117. de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010;376:1147-54.
118. Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med 2013;369:213-23.
119. Wargowski E, Johnson LE, Eickhoff JC, et al. Prime-boost vaccination targeting prostatic acid phosphatase (PAP) in patients with metastatic castration-resistant prostate cancer (mCRPC) using Sipuleucel-T and a DNA vaccine. J Immunother Cancer 2018;6:21.
120. Dannull J, Diener PA, Prikler L, et al. Prostate stem cell antigen is a promising candidate for immunotherapy of advanced prostate cancer. Cancer Res 2000;60:5522-8.
121. Kiessling A, Schmitz M, Stevanovic S, et al. Prostate stem cell antigen: Identification of immunogenic peptides and assessment of reactive CD8+ T cells in prostate cancer patients. Int J Cancer 2002;102:390-7.
122. Matsueda S, Kobayashi K, Nonaka Y, Noguchi M, Itoh K, Harada M. Identification of new prostate stem cell antigen-derived peptides immunogenic in HLA-A2+ patients with hormone-refractory prostate cancer. Cancer Immunol Immunother 2004;53:479-89.
123. Matsueda S, Yao A, Ishihara Y, et al. A prostate stem cell antigen-derived peptide immunogenic in HLA-A24- prostate cancer patients. Prostate 2004;60:205-13.
124. de la Luz Garcia-Hernandez M, Gray A, Hubby B, Klinger OJ, Kast WM. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res 2008;68:861-9.
125. Krupa M, Canamero M, Gomez CE, Najera JL, Gil J, Esteban M. Immunization with recombinant DNA and modified vaccinia virus Ankara (MVA) vectors delivering PSCA and STEAP1 antigens inhibits prostate cancer progression. Vaccine 2011;29:1504-13.
126. Ross S, Spencer SD, Holcomb I, et al. Prostate stem cell antigen as therapy target: tissue expression and in vivo efficacy of an immunoconjugate. Cancer Res 2002;62:2546-53.
127. Saffran DC, Raitano AB, Hubert RS, Witte ON, Reiter RE, Jakobovits A. Anti-PSCA mAbs inhibit tumor growth and metastasis formation and prolong the survival of mice bearing human prostate cancer xenografts. Proc Natl Acad Sci USA 2001;98:2658-63.
128. Gu Z, Yamashiro J, Kono E, Reiter RE. Anti-prostate stem cell antigen monoclonal antibody 1G8 induces cell death in vitro and inhibits tumor growth in vivo via a Fc-independent mechanism. Cancer Res 2005;65:9495-500.
129. Olafsen T, Gu Z, Sherman MA, et al. Targeting, imaging, and therapy using a humanized antiprostate stem cell antigen (PSCA) antibody. J Immunother 2007;30:396-405.
130. Feldmann A, Stamova S, Bippes CC, et al. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: comparison of different antibody formats. Prostate 2011;71:998-1011.
131. Morgenroth A, Cartellieri M, Schmitz M, et al. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate 2007;67:1121-31.
132. Murad JP, Tilakawardane D, Park AK, et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol Ther 2021;29:2335-49.
133. Dorff TB, Narayan V, Forman SJ, et al. Novel redirected T-cell immunotherapies for advanced prostate cancer. Clin Cancer Res 2022;28:576-84.
134. Hempelmann JA, Lockwood CM, Konnick EQ, et al. Microsatellite instability in prostate cancer by PCR or next-generation sequencing. J Immunother Cancer 2018;6:29.
135. Fraune C, Simon R, Höflmayer D, et al. High homogeneity of mismatch repair deficiency in advanced prostate cancer. Virchows Arch 2020;476:745-52.
136. Abida W, Cheng ML, Armenia J, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol 2019;5:471-8.
137. Xu J, Kalos M, Stolk JA, et al. Identification and characterization of prostein, a novel prostate-specific protein. Cancer Res 2001;61:1563-8.
138. Kalos M, Askaa J, Hylander BL, et al. Prostein expression is highly restricted to normal and malignant prostate tissues. Prostate 2004;60:246-56.
139. Musiyenko A, Bitko V, Barik S. Ectopic expression of miR-126*, an intronic product of the vascular endothelial EGF-like 7 gene, regulates prostein translation and invasiveness of prostate cancer LNCaP cells. J Mol Med 2008;86:313-22.
140. Kiessling A, Stevanovic S, Füssel S, et al. Identification of an HLA-A*0201-restricted T-cell epitope derived from the prostate cancer-associated protein prostein. Br J Cancer 2004;90:1034-40.
141. Schmidt U, Fuessel S, Koch R, et al. Quantitative multi-gene expression profiling of primary prostate cancer. Prostate 2006;66:1521-34.
142. Friedman RS, Spies AG, Kalos M. Identification of naturally processed CD8 T cell epitopes from prostein, a prostate tissue-specific vaccine candidate. Eur J Immunol 2004;34:1091-101.
143. Swaya TO, Opondo D, Atandi DO, Guyah B, Magak NWG. Immunohistochemical analysis of prostein in needle core biopsies of acinar and intraductal prostatic adenocarcinoma in Western Kenya population. J Cancer Sci Clin Ther 2022;6:269-75. Available from: https://repository.maseno.ac.ke/handle/123456789/5358 [Last accessed on 12 Apr 2024].
144. Wolfgang CD, Essand M, Vincent JJ, Lee B, Pastan I. TARP: a nuclear protein expressed in prostate and breast cancer cells derived from an alternate reading frame of the T cell receptor gamma chain locus. Proc Natl Acad Sci USA 2000;97:9437-42.
145. Cheng WS, Giandomenico V, Pastan I, Essand M. Characterization of the androgen-regulated prostate-specific T cell receptor gamma-chain alternate reading frame protein (TARP) promoter. Endocrinology 2003;144:3433-40.
146. Maeda H, Nagata S, Wolfgang CD, Bratthauer GL, Bera TK, Pastan I. The T cell receptor gamma chain alternate reading frame protein (TARP), a prostate-specific protein localized in mitochondria. J Biol Chem 2004;279:24561-8.
147. Carlsson B, Tötterman TH, Essand M. Generation of cytotoxic T lymphocytes specific for the prostate and breast tissue antigen TARP. Prostate 2004;61:161-70.
148. Oh S, Terabe M, Pendleton CD, et al. Human CTLs to wild-type and enhanced epitopes of a novel prostate and breast tumor-associated protein, TARP, lyse human breast cancer cells. Cancer Res 2004;64:2610-8.
149. Kobayashi H, Nagato T, Oikawa K, et al. Recognition of prostate and breast tumor cells by helper T lymphocytes specific for a prostate and breast tumor-associated antigen, TARP. Clin Cancer Res 2005;11:3869-78.
150. Epel M, Carmi I, Soueid-Baumgarten S, et al. Targeting TARP, a novel breast and prostate tumor-associated antigen, with T cell receptor-like human recombinant antibodies. Eur J Immunol 2008;38:1706-20.
151. Hillerdal V, Nilsson B, Carlsson B, Eriksson F, Essand M. T cells engineered with a T cell receptor against the prostate antigen TARP specifically kill HLA-A2+ prostate and breast cancer cells. Proc Natl Acad Sci USA 2012;109:15877-81.
152. Hillerdal V, Boura VF, Björkelund H, Andersson K, Essand M. Avidity characterization of genetically engineered T-cells with novel and established approaches. BMC Immunol 2016;17:23.
153. Bonte S, De Munter S, Goetgeluk G, et al. T-cells with a single tumor antigen-specific T-cell receptor can be generated in vitro from clinically relevant stem cell sources. Oncoimmunology 2020;9:1727078.
154. Vanhooren J, Derpoorter C, Depreter B, et al. TARP as antigen in cancer immunotherapy. Cancer Immunol Immunother 2021;70:3061-8.
155. Tsavaler L, Shapero MH, Morkowski S, Laus R. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res 2001;61:3760-9.
156. Ochoa SV, Casas Z, Albarracín SL, Sutachan JJ, Torres YP. Therapeutic potential of TRPM8 channels in cancer treatment. Front Pharmacol 2023;14:1098448.
157. Zhang L, Barritt GJ. TRPM8 in prostate cancer cells: a potential diagnostic and prognostic marker with a secretory function? Endocr Relat Cancer 2006;13:27-38.
158. Bidaux G, Roudbaraki M, Merle C, et al. Evidence for specific TRPM8 expression in human prostate secretory epithelial cells: functional androgen receptor requirement. Endocr Relat Cancer 2005;12:367-82.
159. Valero M, Morenilla-Palao C, Belmonte C, Viana F. Pharmacological and functional properties of TRPM8 channels in prostate tumor cells. Pflugers Arch 2011;461:99-114.
160. Kiessling A, Füssel S, Schmitz M, et al. Identification of an HLA-A*0201-restricted T-cell epitope derived from the prostate cancer-associated protein trp-p8. Prostate 2003;56:270-9.
161. Xu M, Evans L, Bizzaro CL, et al. STEAP1-4 (six-transmembrane epithelial antigen of the prostate 1-4) and their clinical implications for prostate cancer. Cancers 2022;14:4034.
162. Jiao Z, Huang L, Sun J, et al. Six-transmembrane epithelial antigen of the prostate 1 expression promotes ovarian cancer metastasis by aiding progression of epithelial-to-mesenchymal transition. Histochem Cell Biol 2020;154:215-30.
163. Santos C, Socorro S, Maia CJ. STEAP1 (six transmembrane epithelial antigene of the prostate 1). Atlas Genet Cytogenet Oncol Haematol 2011;5:44760.
164. Rodeberg DA, Nuss RA, Elsawa SF, Celis E. Recognition of six-transmembrane epithelial antigen of the prostate-expressing tumor cells by peptide antigen-induced cytotoxic T lymphocytes. Clin Cancer Res 2005;11:4545-52.
165. Alves PM, Faure O, Graff-Dubois S, et al. STEAP, a prostate tumor antigen, is a target of human CD8+ T cells. Cancer Immunol Immunother 2006;55:1515-23.
166. Kobayashi H, Nagato T, Sato K, et al. Recognition of prostate and melanoma tumor cells by six-transmembrane epithelial antigen of prostate-specific helper T lymphocytes in a human leukocyte antigen class II-restricted manner. Cancer Res 2007;67:5498-504.
167. Azumi M, Kobayashi H, Aoki N, et al. Six-transmembrane epithelial antigen of the prostate as an immunotherapeutic target for renal cell and bladder cancer. J Urol 2010;183:2036-44.
168. Hayashi S, Kumai T, Matsuda Y, et al. Six-transmembrane epithelial antigen of the prostate and enhancer of zeste homolog 2 as immunotherapeutic targets for lung cancer. J Transl Med 2011;9:191.
169. de la Luz Garcia-Hernandez M, Gray A, Hubby B, Kast WM. In vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: a candidate antigen for treating prostate cancer. Cancer Res 2007;67:1344-51.
170. Kim S, Lee JB, Lee GK, Chang J. Vaccination with recombinant adenoviruses and dendritic cells expressing prostate-specific antigens is effective in eliciting CTL and suppresses tumor growth in the experimental prostate cancer. Prostate 2009;69:938-48.
171. Challita-Eid PM, Morrison K, Etessami S, et al. Monoclonal antibodies to six-transmembrane epithelial antigen of the prostate-1 inhibit intercellular communication in vitro and growth of human tumor xenografts in vivo. Cancer Res 2007;67:5798-805.
172. Gati A, Lajmi N, Derouiche A, Marrakchi R, Chebil M, Benammar-Elgaaied A. NY-ESO-1 expression and immunogenicity in prostate cancer patients. Tunisie Med 2011;89:779-83.
173. Thomas R, Al-Khadairi G, Roelands J, et al. NY-ESO-1 based immunotherapy of cancer: current perspectives. Front Immunol 2018;9:947.
174. Nakada T, Noguchi Y, Satoh S, et al. NY-ESO-1 mRNA expression and immunogenicity in advanced prostate cancer. Cancer Immun 2003;3:10.
175. Karbach J, Neumann A, Atmaca A, et al. Efficient in vivo priming by vaccination with recombinant NY-ESO-1 protein and CpG in antigen naive prostate cancer patients. Clin Cancer Res 2011;17:861-70.
177. Arima Y, Matsueda S, Yano H, Harada M, Itoh K. Parathyroid hormone-related protein as a common target molecule in specific immunotherapy for a wide variety of tumor types. Int J Oncol 2005;27:981-8.
178. Edwards CM, Johnson RW. From good to bad: the opposing effects of PTHrP on tumor growth, dormancy, and metastasis throughout cancer progression. Front Oncol 2021;11:644303.
179. Asadi F, Kukreja S. Parathyroid hormone-related protein in prostate cancer. Crit Rev Eukaryot Gene Expr 2005;15:15-28.
180. Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994;266:2011-5.
181. Adotévi O, Mollier K, Neuveut C, et al. Immunogenic HLA-B*0702-restricted epitopes derived from human telomerase reverse transcriptase that elicit antitumor cytotoxic T-cell responses. Clin Cancer Res 2006;12:3158-67.
182. Negrini S, De Palma R, Filaci G. Anti-cancer immunotherapies targeting telomerase. Cancers 2020;12:2260.
183. Huo LF, Tang JW, Huang JJ, et al. Cancer immunotherapy targeting the telomerase reverse transcriptase. Cell Mol Immunol 2006;3:1-11.
184. Leão R, Apolónio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: clinical impacts in cancer. J Biomed Sci 2018;25:22.
185. Esquenazi Y, Ballester LY. Telomerase reverse transcriptase alterations in human cancers: diagnosis, prognosis, and therapeutic implications. Cancer Cytopathol 2019;127:275-7.
186. Yang R, Han Y, Guan X, et al. Regulation and clinical potential of telomerase reverse transcriptase (TERT/hTERT) in breast cancer. Cell Commun Signal 2023;21:218.
187. Ryan BM, O’Donovan N, Duffy MJ. Survivin: a new target for anti-cancer therapy. Cancer Treat Rev 2009;35:553-62.
188. Zhang M, Latham DE, Delaney MA, Chakravarti A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene 2005;24:2474-82.
189. Xiang R, Mizutani N, Luo Y, et al. A DNA vaccine targeting survivin combines apoptosis with suppression of angiogenesis in lung tumor eradication. Cancer Res 2005;65:553-61.
190. Ciesielski MJ, Kozbor D, Castanaro CA, Barone TA, Fenstermaker RA. Therapeutic effect of a T helper cell supported CTL response induced by a survivin peptide vaccine against murine cerebral glioma. Cancer Immunol Immunother 2008;57:1827-35.
191. Li F, Aljahdali I, Ling X. Cancer therapeutics using survivin BIRC5 as a target: what can we do after over two decades of study? J Exp Clin Cancer Res 2019;38:368.
192. Garg H, Suri P, Gupta JC, Talwar GP, Dubey S. Survivin: a unique target for tumor therapy. Cancer Cell Int 2016;16:49.
193. Di Lorenzo G, Autorino R, De Laurentiis M, et al. HER-2/neu receptor in prostate cancer development and progression to androgen independence. Tumori 2004;90:163-70.
194. Neto AS, Tobias-Machado M, Wroclawski ML, et al. Her-2/neu expression in prostate adenocarcinoma: a systematic review and meta-analysis. J Urol 2010;184:842-50.
195. Baxevanis CN, Voutsas IF, Gritzapis AD, Perez SA, Papamichail M. HER-2/neu as a target for cancer vaccines. Immunotherapy 2010;2:213-26.
196. Agus DB, Scher HI, Higgins B, et al. Response of prostate cancer to anti-Her-2/neu antibody in androgen-dependent and -independent human xenograft models. Cancer Res 1999;59:4761-4.
197. Formento P, Hannoun-Levi JM, Gérard F, et al. Gefitinib-trastuzumab combination on hormone-refractory prostate cancer xenograft. Eur J Cancer 2005;41:1467-73.
198. Pinthus JH, Waks T, Malina V, et al. Adoptive immunotherapy of prostate cancer bone lesions using redirected effector lymphocytes. J Clin Invest 2004;114:1774-81.
199. De Muga S, Hernández S, Agell L, et al. Molecular alterations of EGFR and PTEN in prostate cancer: association with high-grade and advanced-stage carcinomas. Mod Pathol 2010;23:703-12.
200. Filho PA, López-Albaitero A, Gooding W, Ferris RL. Novel immunogenic HLA-A*0201-restricted epidermal growth factor receptor-specific T-cell epitope in head and neck cancer patients. J Immunother 2010;33:83-91.
201. Vexler A, Lidawi G, Loew V, et al. Anti-ErbB4 targeting approaches for prostate cancer treatment. Cancer Biol Ther 2008;7:1090-4.
202. Richards L. Genetics: N-cadherin - a target for prostate cancer therapy. Nat Rev Clin Oncol 2011;8:63.
203. Tanaka H, Kono E, Tran CP, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med 2010;16:1414-20.
204. Cui Y, Yamada S. N-cadherin dependent collective cell invasion of prostate cancer cells is regulated by the N-terminus of α-catenin. PLoS One 2013;8:e55069.
205. Jennbacken K, Tesan T, Wang W, Gustavsson H, Damber JE, Welén K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr Relat Cancer 2010;17:469-79.
206. Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets 2011;15:31-51.
207. Colapietro A, Gravina GL, Petragnano F, et al. Antitumorigenic effects of inhibiting ephrin receptor kinase signaling by GLPG1790 against colorectal cancer cell lines in vitro and in vivo. J Oncol 2020;2020:9342732.
208. Chang FL, Tsai KC, Lin TY, Chiang CW, Pan SL, Lee YC. Effectiveness of anti-erythropoietin producing hepatocellular receptor Type-A2 antibody in pancreatic cancer treatment. Heliyon 2023;9:e21774.
209. Yamaguchi S, Tatsumi T, Takehara T, et al. Immunotherapy of murine colon cancer using receptor tyrosine kinase EphA2-derived peptide-pulsed dendritic cell vaccines. Cancer 2007;110:1469-77.
210. Peng L, Oganesyan V, Damschroder MM, Wu H, Dall’Acqua WF. Structural and functional characterization of an agonistic anti-human EphA2 monoclonal antibody. J Mol Biol 2011;413:390-405.
211. Lee JW, Stone RL, Lee SJ, et al. EphA2 targeted chemotherapy using an antibody drug conjugate in endometrial carcinoma. Clin Cancer Res 2010;16:2562-70.
212. Smith HA, Cronk RJ, Lang JM, McNeel DG. Expression and immunotherapeutic targeting of the SSX family of cancer-testis antigens in prostate cancer. Cancer Res 2011;71:6785-95.
213. Hale R, Sandakly S, Shipley J, Walters Z. Epigenetic targets in synovial sarcoma: a mini-review. Front Oncol 2019;9:1078.
214. Smith HA, McNeel DG. The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin Dev Immunol 2010;2010:150591.
215. Smith HA, McNeel DG. Vaccines targeting the cancer-testis antigen SSX-2 elicit HLA-A2 epitope-specific cytolytic T cells. J Immunother 2011;34:569-80.
216. Liao Y, Wu M, Jia Y, Mou R, Li X. EpCAM as a novel biomarker for survivals in prostate cancer patients. Front Cell Dev Biol 2022;10:843604.
217. Ni J, Cozzi P, Beretov J, et al. Epithelial cell adhesion molecule (EpCAM) is involved in prostate cancer chemotherapy/radiotherapy response in vivo. BMC Cancer 2018;18:1092.
218. Rybalov M, Ananias HJ, Hoving HD, van der Poel HG, Rosati S, de Jong IJ. PSMA, EpCAM, VEGF and GRPR as imaging targets in locally recurrent prostate cancer after radiotherapy. Int J Mol Sci 2014;15:6046-61.
219. Yan Y, Zhou B, Qian C, et al. RIPK2 stabilizes c-Myc and is an actionable target for inhibiting prostate cancer metastasis. bioRxiv 2020.
220. Yan Y, Zhou B, Qian C, et al. Receptor-interacting protein kinase 2 (RIPK2) stabilizes c-Myc and is a therapeutic target in prostate cancer metastasis. Nat Commun 2022;13:669.
221. You J, Wang Y, Chen H, Jin F. RIPK2: a promising target for cancer treatment. Front Pharmacol 2023;14:1192970.
222. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019;18:197-218.
223. Karpf AR, Bai S, James SR, Mohler JL, Wilson EM. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol Cancer Res 2009;7:523-35.
224. Bai S, Wilson EM. Epidermal-growth-factor-dependent phosphorylation and ubiquitinylation of MAGE-11 regulates its interaction with the androgen receptor. Mol Cell Biol 2008;28:1947-63.
225. Su S, Parris AB, Grossman G, Mohler JL, Wang Z, Wilson EM. Up-regulation of follistatin-like 1 by the androgen receptor and melanoma Antigen-A11 in prostate cancer. Prostate 2017;77:505-16.
226. Bai S, He B, Wilson EM. Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol Cell Biol 2005;25:1238-57.
228. Gasser S, Lim LHK, Cheung FSG. The role of the tumour microenvironment in immunotherapy. Endocr Relat Cancer 2017;24:T283-95.
229. Stultz J, Fong L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis 2021;24:697-717.
230. Wu Z, Chen H, Luo W, et al. The landscape of immune cells infiltrating in prostate cancer. Front Oncol 2020;10:517637.
231. Di Mitri D, Mirenda M, Vasilevska J, et al. Re-education of tumor-associated macrophages by CXCR2 blockade drives senescence and tumor inhibition in advanced prostate cancer. Cell Rep 2019;28:2156-68.e5.
232. Ollivier L, Labbé M, Fradin D, Potiron V, Supiot S. Interaction between modern radiotherapy and immunotherapy for metastatic prostate cancer. Front Oncol 2021;11:744679.
233. Hess KR, Varadhachary GR, Taylor SH, et al. Metastatic patterns in adenocarcinoma. Cancer 2006;106:1624-33.
234. Xiang L, Gilkes DM. The contribution of the immune system in bone metastasis pathogenesis. Int J Mol Sci 2019;20:999.
235. Kim SW, Kim JS, Papadopoulos J, et al. Consistent interactions between tumor cell IL-6 and macrophage TNF-α enhance the growth of human prostate cancer cells in the bone of nude mouse. Int Immunopharmacol 2011;11:862-72.
236. Jiao S, Subudhi SK, Aparicio A, et al. Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell 2019;179:1177-90.e13.
237. Tan Y, Wang M, Zhang Y, et al. Tumor-associated macrophages: a potential target for cancer therapy. Front Oncol 2021;11:693517.
238. Kumari N, Choi SH. Tumor-associated macrophages in cancer: recent advancements in cancer nanoimmunotherapies. J Exp Clin Cancer Res 2022;41:68.
240. Ioannidou E, Moschetta M, Shah S, et al. Angiogenesis and anti-angiogenic treatment in prostate cancer: mechanisms of action and molecular targets. Int J Mol Sci 2021;22:9926.
241. Sharma V, Dong H, Kwon E, Karnes RJ. Positive pelvic lymph nodes in prostate cancer harbor immune suppressor cells to impair tumor-reactive T cells. Eur Urol Focus 2018;4:75-9.
242. Chen S, Zhu G, Yang Y, et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat Cell Biol 2021;23:87-98.
243. Klusa D, Lohaus F, Furesi G, et al. Metastatic spread in prostate cancer patients influencing radiotherapy response. Front Oncol 2020;10:627379.
244. He ZN, Zhang CY, Zhao YW, et al. Regulation of T cells by myeloid-derived suppressor cells: emerging immunosuppressor in lung cancer. Discov Oncol 2023;14:185.
245. Venkatachalam S, McFarland TR, Agarwal N, Swami U. Immune checkpoint inhibitors in prostate cancer. Cancers 2021;13:2187.
246. Gonzalez-Velez M, Bryce A. How to approach immune checkpoint inhibitor therapy in prostate cancer. Urol Times Urologists Cancer Care 2020;9. Available from: https://www.urologytimes.com/view/how-to-approach-immune-checkpoint-inhibitor-therapy-in-prostate-cancer [Last accessed on 29 Mar 2024]
247. Wong YNS, Sankey P, Josephs DH, et al. Nivolumab and ipilimumab treatment in prostate cancer with an immunogenic signature (NEPTUNES). J Clin Oncol 2019;37:tps5090.
248. Carbognin L, Pilotto S, Milella M, et al. Differential activity of nivolumab, pembrolizumab and MPDL3280A according to the tumor expression of programmed death-ligand-1 (PD-L1): sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS One 2015;10:e0130142.
249. Sato H, Niimi A, Yasuhara T, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 2017;8:1751.
250. Subudhi SK, Vence L, Zhao H, et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci Transl Med 2020;12:eaaz3577.
251. Beer TM, Kwon ED, Drake CG, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 2017;35:40-7.
252. Kwon ED, Drake CG, Scher HI, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 2014;15:700-12.
253. Graff JN, Burgents J, Liang LW, Stenzl A. Phase III study of pembrolizumab (Pembro) plus enzalutamide (Enza) versus placebo plus enza for metastatic castration-resistant prostate cancer (mCRPC): KEYNOTE-641. J Clin Oncol 2020;38:TPS258.
254. Antonarakis ES, Park SH, Goh JC, et al. Pembrolizumab plus olaparib for patients with previously treated and biomarker-unselected metastatic castration-resistant prostate cancer: the randomized, open-label, phase III KEYLYNK-010 trial. J Clin Oncol 2023;41:3839-50.
255. Petrylak DP, Ratta R, Matsubara N, et al. Pembrolizumab plus docetaxel for patients with metastatic castration-resistant prostate cancer (mCRPC): randomized, double-blind, phase 3 KEYNOTE-921 study. J Clin Oncol 2023;41:19.
256. Powles T, Yuen KC, Gillessen S, et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: a randomized phase 3 trial. Nat Med 2022;28:144-53.
257. Fizazi K, González Mella P, Castellano D, et al. Nivolumab plus docetaxel in patients with chemotherapy-naïve metastatic castration-resistant prostate cancer: results from the phase II CheckMate 9KD trial. Eur J Cancer 2022;160:61-71.
258. Meng L, Yang Y, Mortazavi A, Zhang J. Emerging immunotherapy approaches for treating prostate cancer. Int J Mol Sci 2023;24:14347.
259. Hansen AR, Massard C, Ott PA, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann Oncol 2018;29:1807-13.
260. Antonarakis ES, Piulats JM, Gross-Goupil M, et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II KEYNOTE-199 study. J Clin Oncol 2020;38:395-405.
261. Velho P, Antonarakis ES. PD-1/PD-L1 pathway inhibitors in advanced prostate cancer. Expert Rev Clin Pharmacol 2018;11:475-86.
262. Wang S, Xie K, Liu T. Cancer immunotherapies: from efficacy to resistance mechanisms - not only checkpoint matters. Front Immunol 2021;12:690112.
263. Liang H, Liu Y, Guo J, et al. Progression in immunotherapy for advanced prostate cancer. Front Oncol 2023;13:1126752.
265. Lorenzo G, Buonerba C, Kantoff PW. Immunotherapy for the treatment of prostate cancer. Nat Rev Clin Oncol 2011;8:551-61.
266. Galsky MD. Resistance to prostate-cancer treatment is driven by immune cells. Nature 2018;559:338-9.
267. Belete TM. The current status of gene therapy for the treatment of cancer. Biologics 2021;15:67-77.
268. Edwards BK, Noone AM, Mariotto AB, et al. Annual report to the nation on the status of cancer, 1975-2010, featuring prevalence of comorbidity and impact on survival among persons with lung, colorectal, breast, or prostate cancer. Cancer 2014;120:1290-314.
269. National Cancer Institute. NCI comorbidity index overview. Available from: https://healthcaredelivery.cancer.gov/seermedicare/considerations/comorbidity.html [Last accessed on 28 Mar 2024].
270. Klabunde CN, Potosky AL, Legler JM, Warren JL. Development of a comorbidity index using physician claims data. J Clin Epidemiol 2000;53:1258-67.
271. Asmar R, Beebe-Dimmer JL, Korgavkar K, Keele GR, Cooney KA. Hypertension, obesity and prostate cancer biochemical recurrence after radical prostatectomy. Prostate Cancer Prostatic Dis 2013;16:62-6.
272. Post JM, Beebe-Dimmer JL, Morgenstern H, et al. The metabolic syndrome and biochemical recurrence following radical prostatectomy. Prostate Cancer 2011;2011:245642.
273. Jefferson M, Drake RR, Lilly M, Savage SJ, Tucker Price S, Hughes Halbert C. Co-morbidities in a retrospective cohort of prostate cancer patients. Ethn Dis 2020;30:185-92.
274. Boussios S, Rassy E, Samartzis E, et al. Melanoma of unknown primary: new perspectives for an old story. Crit Rev Oncol Hematol 2021;158:103208.
275. Sharma AR, Kundu SK, Nam JS, et al. Next generation delivery system for proteins and genes of therapeutic purpose: why and how? Biomed Res Int 2014;2014:327950.
276. Templeton NS, editor. Gene and cell therapy: therapeutic mechanisms and strategies. CRC Press; 2008.
277. Colella P, Ronzitti G, Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther Methods Clin Dev 2018;8:87-104.
278. Merten OW, Charrier S, Laroudie N, et al. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther 2011;22:343-56.
279. Huang C, Li G, Wu J, Liang J, Wang X. Identification of pathogenic variants in cancer genes using base editing screens with editing efficiency correction. Genome Biol 2021;22:80.
280. Zhang H, Qin C, An C, et al. Application of the CRISPR/Cas9-based gene editing technique in basic research, diagnosis, and therapy of cancer. Mol Cancer 2021;20:126.
281. Palpant NJ, Dudzinski D. Zinc finger nucleases: looking toward translation. Gene Ther 2013;20:121-7.
282. Cassandri M, Smirnov A, Novelli F, et al. Zinc-finger proteins in health and disease. Cell Death Discov 2017;3:17071.
283. Shojaei Baghini S, Gardanova ZR, Abadi SAH, et al. CRISPR/Cas9 application in cancer therapy: a pioneering genome editing tool. Cell Mol Biol Lett 2022;27:35.
284. Hille F, Richter H, Wong SP, Bratovič M, Ressel S, Charpentier E. The biology of CRISPR-Cas: backward and forward. Cell 2018;172:1239-59.
285. Li Y, Glass Z, Huang M, Chen ZY, Xu Q. Ex vivo cell-based CRISPR/Cas9 genome editing for therapeutic applications. Biomaterials 2020;234:119711.
286. Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol Bioeng 2013;110:1811-21.
287. Nakano C, Kitabatake Y, Takeyari S, et al. Genetic correction of induced pluripotent stem cells mediated by transcription activator-like effector nucleases targeting ALPL recovers enzyme activity and calcification in vitro. Mol Genet Metab 2019;127:158-65.
289. Bin Umair M, Akusa FN, Kashif H, et al. Viruses as tools in gene therapy, vaccine development, and cancer treatment. Arch Virol 2022;167:1387-404.
290. Sung YK, Kim SW. Recent advances in the development of gene delivery systems. Biomater Res 2019;23:8.
291. Sung YK, Kim SW. The practical application of gene vectors in cancer therapy. Integrat Cancer Sci Therap 2018;5:1-5.
292. Panday R, Abdalla AM, Neupane M, Khadka S, Kricha A, Yang G. Advances in magnetic nanoparticle-driven delivery of gene therapies towards prostate cancer. J Nanomater 2021;2021:6050795.
293. Altwaijry N, Somani S, Dufès C. Targeted nonviral gene therapy in prostate cancer. Int J Nanomed 2018;13:5753-67.
294. Rehman K, Iqbal Z, Zhiqin D, et al. Analysis of genetic biomarkers, polymorphisms in ADME-related genes and their impact on pharmacotherapy for prostate cancer. Cancer Cell Int 2023;23:247.
295. Ahmed KA, Davis BJ, Wilson TM, Wiseman GA, Federspiel MJ, Morris JC. Progress in gene therapy for prostate cancer. Front Oncol 2012;2:172.
296. Naseer F, Ahmad T, Kousar K, Anjum S. Advanced therapeutic options for treatment of metastatic castration resistant prostatic adenocarcinoma. Front Pharmacol 2021;12:728054.
297. Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discov 2017;16:387-99.
299. Hill AB, Chen M, Chen CK, Pfeifer BA, Jones CH. Overcoming gene-delivery hurdles: physiological considerations for nonviral vectors. Trends Biotechnol 2016;34:91-105.
300. Zhang Y, Satterlee A, Huang L. In vivo gene delivery by nonviral vectors: overcoming hurdles? Mol Ther 2012;20:1298-304.
301. Butt MH, Zaman M, Ahmad A, et al. Appraisal for the potential of viral and nonviral vectors in gene therapy: a review. Genes 2022;13:1370.
302. Dey D, Evans GR. Suicide gene therapy by herpes simplex virus-1 thymidine kinase (HSV-TK). Targets Gene Ther 2011:65.
303. Harrison GS, Glode LM. Current challenges of gene therapy for prostate cancer. Oncology 1997;11:845-55.
304. Han C, Deng Y, Xu W, et al. The roles of tumor-associated macrophages in prostate cancer. J Oncol 2022;2022:8580043.
305. Msaouel P, Iankov ID, Allen C, et al. Engineered measles virus as a novel oncolytic therapy against prostate cancer. Prostate 2009;69:82-91.
306. Li J, Røise JJ, He M, Das R, Murthy N. Non-viral strategies for delivering genome editing enzymes. Adv Drug Deliv Rev 2021;168:99-117.
307. Gregg JR, Thompson TC. Considering the potential for gene-based therapy in prostate cancer. Nat Rev Urol 2021;18:170-84.
308. van Ophoven A, Ng CP, Patel B, Bonavida B, Belldegrun A. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) for treatment of prostate cancer: first results and review of the literature. Prostate Cancer Prostatic Dis 1999;2:227-33.
309. Darvish L, Bahreyni-Toossi MT, Aghaee-Bakhtiari SH, et al. Inducing apoptosis by using microRNA in radio-resistant prostate cancer: an in-silico study with an in-vitro validation. Mol Biol Rep 2023;50:6063-74.
310. Teh BS, Ishiyama H, Mai WY, Thompson TC, Butler EB. Long-term outcome of a phase II trial using immunomodulatory in situ gene therapy in combination with intensity-modulated radiotherapy with or without hormonal therapy in the treatment of prostate cancer. Int J Radiat Oncol 2015;4:377-86.
311. Boettcher AN, Usman A, Morgans A, VanderWeele DJ, Sosman J, Wu JD. Past, current, and future of immunotherapies for prostate cancer. Front Oncol 2019;9:884.
312. LeVee A, Lin CY, Posadas E, et al. Clinical utility of olaparib in the treatment of metastatic castration-resistant prostate cancer: a review of current evidence and patient selection. Onco Targets Ther 2021;14:4819-32.
313. Powers E, Karachaliou GS, Kao C, et al. Novel therapies are changing treatment paradigms in metastatic prostate cancer. J Hematol Oncol 2020;13:144.
314. Gdor Y, Timme TL, Kadmon D, Miles BJ, Thompson TC. Strategies for prostate cancer gene therapy. Am J Cancer 2004;3:79-95.
315. Lin Y, Wagner E, Lächelt U. Non-viral delivery of the CRISPR/Cas system: DNA versus RNA versus RNP. Biomater Sci 2022;10:1166-92.
316. Kanvinde S, Kulkarni T, Deodhar S, Bhattacharya D, Dasgupta A. Non-viral vectors for delivery of nucleic acid therapies for cancer. BioTech 2022;11:6.
317. McCrudden CM, McBride JW, McCaffrey J, et al. Gene therapy with RALA/iNOS composite nanoparticles significantly enhances survival in a model of metastatic prostate cancer. Cancer Nanotechnol 2018;9:5.
318. Liu C, Hasegawa K, Russell SJ, Sadelain M, Peng KW. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate 2009;69:1128-41.
319. Son HA, Zhang L, Cuong BK, et al. Combination of vaccine-strain measles and mumps viruses enhances oncolytic activity against human solid malignancies. Cancer Invest 2018;36:106-17.
320. Durso RJ, Andjelic S, Gardner JP, et al. A novel alphavirus vaccine encoding prostate-specific membrane antigen elicits potent cellular and humoral immune responses. Clin Cancer Res 2007;13:3999-4008.
321. Mansfield DC, Kyula JN, Rosenfelder N, et al. Oncolytic vaccinia virus as a vector for therapeutic sodium iodide symporter gene therapy in prostate cancer. Gene Ther 2016;23:357-68.
322. Slovin SF, Kehoe M, Durso R, et al. A phase I dose escalation trial of vaccine replicon particles (VRP) expressing prostate-specific membrane antigen (PSMA) in subjects with prostate cancer. Vaccine 2013;31:943-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Bibi R, Sarkar K. New approaches and prospects of immunotherapy and gene therapy for prostate cancer. J Transl Genet Genom 2024;8:119-61. http://dx.doi.org/10.20517/jtgg.2023.50
AMA Style
Bibi R, Sarkar K. New approaches and prospects of immunotherapy and gene therapy for prostate cancer. Journal of Translational Genetics and Genomics. 2024; 8(2): 119-61. http://dx.doi.org/10.20517/jtgg.2023.50
Chicago/Turabian Style
Bibi, Roshni, Koustav Sarkar. 2024. "New approaches and prospects of immunotherapy and gene therapy for prostate cancer" Journal of Translational Genetics and Genomics. 8, no.2: 119-61. http://dx.doi.org/10.20517/jtgg.2023.50
ACS Style
Bibi, R.; Sarkar K. New approaches and prospects of immunotherapy and gene therapy for prostate cancer. J. Transl. Genet. Genom. 2024, 8, 119-61. http://dx.doi.org/10.20517/jtgg.2023.50
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 0 clicks
Cite This Article 0 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.