
Clinical characterization of a new individual with mild SC4MOL deficiency: diagnostic and therapeutic implications

Abstract
Sterol C4-methyloxidase-like (SC4MOL) deficiency is an autosomal recessive condition caused by biallelic pathogenic variants in MSMO1, resulting in the accumulation of 4-monomethyl and 4,4′-dimethyl sterols due to an enzymatic block in the cholesterol synthesis pathway. SC4MOL deficiency was first reported in 2011, with only seven additional cases from five unrelated families described in the literature since. Based on these reports, the most characteristic clinical features include the triad of microcephaly, congenital cataracts, and psoriatic dermatitis, followed by delayed growth and puberty, and neurodevelopmental problems. Herein, we describe an 8-year-old boy who presented with congenital cataracts and developmental delay at age 6 months and was found to have biallelic variants in MSMO1 by trio exome sequencing. Initial total methylsterol levels were elevated but responsive to statin therapy, while total cholesterol levels remained normal throughout. Available clinical and biochemical data suggest this individual could represent the mildest case of SC4MOL deficiency to date.
Keywords
INTRODUCTION
Cholesterol is a complex sterol molecule that participates in multiple cellular processes, including cell wall permeability/stability, mitosis and meiosis, Hedgehog signaling, and ultimately embryonic development[1,2]. Dietary intake of cholesterol is responsible for approximately 25% of serum cholesterol, with the remainder synthesized de novo from acetyl-CoA primarily in the liver[3]. The de novo cholesterol biosynthesis pathway requires more than 20 enzymes and is generally divided into two components: pre-squalene and post-squalene cholesterol synthesis[4]. Pre-squalene synthesis leads to the production of squalene, used by post-squalene synthesis for sterol and vitamin D production[4]. To date, 17 separate genetic diseases have been described due to single enzyme deficiencies in the de novo cholesterol synthesis pathway, with 10 of the 17 representing a disorder of post-squalene synthesis[5].
The sterol C4-methyloxidase like (SC4MOL) or methylsterol monooxygenase 1 (MSMO1) enzyme functions within the sterol demethylation complex of the post-squalene synthesis pathway, catalyzing the first step in demethylation of 4,4′-dimethylsterols[5]. SC4MOL deficiency (OMIM #616834) is an autosomal recessive genetic condition caused by biallelic pathogenic variants in the gene, MSMO1, resulting in the accumulation of 4-monomethyl and 4,4′-dimethyl sterols[6,7]. These methylsterols are also referred to as meiosis-activating sterols (MASs) based on their role in mammalian meiosis[8,9]. Although there is no clear evidence of their effect on mitosis and cell cycle regulation, there is documentation of skin cell hyperproliferation in individuals with this disorder, suggesting a higher rate of cell division[7]. Other effects of MASs accumulation include immune dysregulation such as overactivation of CD16+ and toll-like receptor (TLR)-2+ granulocytes (potentially leading to an abnormal response in presence of bacterial infections), reduced endothelial-growth factor receptor signal transduction, and impaired intracellular vesicular trafficking[6].
The first individual with SC4MOL deficiency was described in 2011; only seven additional cases from five unrelated families have been reported since[6,7,10,11]. Based on these reports, the most characteristic clinical features of SC4MOL deficiency include the triad of microcephaly, congenital cataracts, and psoriatic dermatitis (MCCPD), followed by delayed growth and puberty, developmental delay, and intellectual disability. Reported biochemical abnormalities consist of increased levels of 4-monomethyl and
Herein, we describe an 8-year-old boy who presented with congenital cataracts and moderate developmental delay at 6 months of age, who was found to have a likely pathogenic variant and a variant of uncertain significance (VUS) in MSMO1 by trio exome sequencing. Initial total methylsterol levels were elevated, but decreased as statin therapy was implemented with a positive impact on his clinical status.
CASE REPORT
A 6-month-old Caucasian boy presented for evaluation by medical genetics due to a concern for congenital cataracts and developmental delay [Figure 1]. There were no prenatal complications other than advanced maternal age. He was born at 39 weeks of gestation by C-section due to failure to progress to a 35-year-old mother and father. His birth growth parameters were weight 3.5 kg (48th centile), length 51 cm (65th centile), and head circumference 35.4 cm (41st centile). Shortly after birth, he was identified with left lacrimal obstruction requiring observation. As he grew older, his parents noticed eye misalignment, which by 6 months raised the concern for bilateral congenital cataracts due to absent red reflexes. Cataracts were confirmed and lens removal was performed at 7 months of age. Family history consisted of Italian, German, and Basque ancestry, but was otherwise non-contributory.
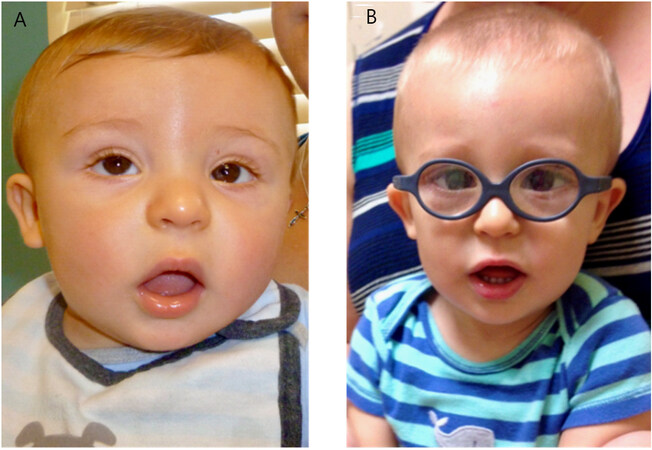
Figure 1. Selected facial images of proband (Case 8). (A) At age 6 months. (B) At age 18 months.
During infancy, he exhibited signs of developmental delay with decreased head control and inability to sit by 6 months of age. He was placed in physical, speech, and occupational therapy. Developmental milestones included sitting independently at 10 months, standing with support at 12 months, walking independently by 18 months, and speaking his first word at 11 months. His speech was quite limited until after age 2 years when he started to communicate by using multiple words.
While an infant, he also exhibited intermittent perianal redness that showed mild response to over-the-counter topical ointments or prescribed topical steroids.
At 14 months, growth parameters included weight 8.90 kg (3rd centile), height 77.5 cm (46th centile) and head circumference 45.7 cm (17th centile). Initial physical exam was remarkable for nystagmus, right-side thoracic depression, prominent left-side rib margins, and bilateral fifth finger clinodactyly.
Comprehensive metabolic panel, creatine kinase, echocardiogram, acylcarnitine profile, and oligo-SNP array were all normal.
At 27 months, trio exome sequencing revealed biallelic variants in MSMO1, consisting of a maternally inherited VUS, c.536C>T (p.P179L), and a paternally inherited likely pathogenic variant, c.731A>G (p.Y244C). Upon identification of these variants, plasma sterol analysis revealed a normal cholesterol level and normal levels of proximal cholesterol precursors (desmosterol, 7-dehydrocholesterol, and lathosterol) but increased levels of 4-monomethyl and 4,4′-dimethyl sterols in a pattern consistent with SC4MOL deficiency [Figure 2]. He was started on simvastatin (0.25 mg/kg/d) to inhibit hydroxymethylglutaryl-CoA (HMG-CoA) reductase and stimulate any residual SC4MOL enzyme activity, and coenzyme Q10 (CoQ10) (50 mg/d), to reduce potential adverse effects from simvastatin intake. However, methylsterol levels after 3 months of statin therapy (30 months of age) remained increased [Figure 2], requiring a simvastatin dose increase to 0.5 mg/kg/d.
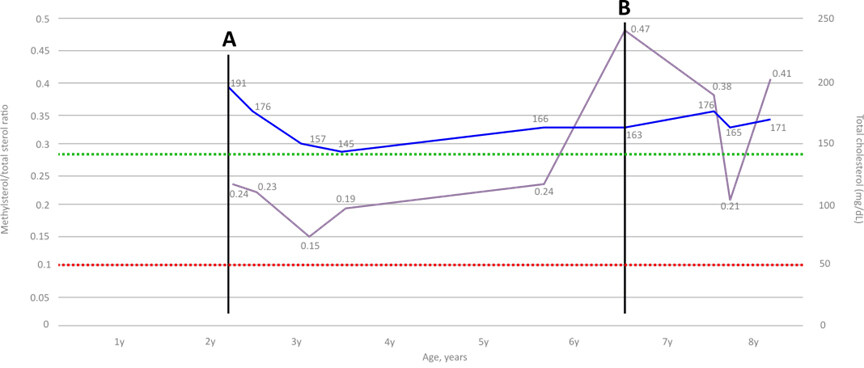
Figure 2. Total cholesterol and methylsterol/total sterol ratio through proband’s life. (A) Simvastatin and CoQ10 were initiated at time of diagnosis. (B) Pravastatin added at 6.4 years of age, after an increase in methylsterol levels. Purple line: methylsterol/total sterol ratios. Blue line:total cholesterol levels. Red dotted line:normal methylsterol/total sterol ratio (< 0.1%). Green dotted line: lower normal limit of total cholesterol (< 140 mg/dL).
Six months after initiating statin therapy (age 34 months), marked improvements in his language were noted, including the ability to say 25-50 words and communicate in three-word sentences. A follow-up methysterol level analysis at 37 months showed a decrease in total methylsterol levels with a reduced methylsterol/total sterol ratio of 0.15 [Figure 2].
Clinical adverse events to simvastatin therapy have been limited to myalgia. Statin-related monitoring of transaminases, creatine kinase, and renal function has remained stable and within normal limits to date. Total cholesterol, high-density lipoprotein (HDL), triglycerides, and low-density lipoprotein (LDL) surveillance were initiated at 53 months of age, with no major alterations except for elevated triglycerides (104 mg/dL) at 6 years 5 months of age. This, along with rising methylsterol levels, prompted the addition of pravastatin to his statin regimen [Figure 2]. Triglycerides normalized (49 mg/dl) by age 8 years.
At age 7 years, the proband was diagnosed with autism spectrum disorder, obsessive-compulsive disorder, and anxiety, requiring dexmethylphenidate. He exhibited a severe perianal rash with excoriation and other ichthyotic skin changes that responded only to a topical combination of cholesterol 10% and simvastatin 2%. He was also diagnosed with mild bilateral genu valgum, requiring orthotics.
Currently, at the age of 8 years 2 months, his growth parameters are in the normal range, with height 129 cm (48th centile), weight 35.3 kg (93rd centile) and head circumference 52.5 cm (47th centile). His physical exam continues to be unremarkable with resolution of his severe prior perianal inflammation. He attends elementary school where he receives special education in addition to physical and occupational therapy. His most recent sterol studies continue to show a normal cholesterol level, but there has been a slight upward trend in the methylsterol/total sterol ratio [Figure 2]. His current therapeutic regimen consists of simvastatin 10 mg/day (0.3 mg/kg/d), pravastatin 4 mg/day, and CoQ10 50 mg/day. He is followed by endocrinology because of the risk of potential cholesterol-dependent hormone deficiencies. His last evaluation by ophthalmology did not show evidence of additional complications, and there are plans for lens implantation between ages 10 to 12 years.
DISCUSSION
SC4MOL deficiency is considered to be one of the rarest cholesterol synthesis disorders with only seven cases from five unrelated families described to date[6,7,10,11]. The most common clinical findings in affected individuals include early-childhood onset of psoriasiform dermatitis or dry skin (n = 6), neurodevelopmental problems (n = 5), microcephaly (n = 4), growth delay (n = 4), congenital cataracts
Clinical features of previously and currently reported patients with SC4MOL deficiency
| He et al.[6,7] 2011 & 2014 | Frisso et al.[10] 2017 | Kalay et al.[11] 2020 | Morales et al. 2022 | |||||
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | |
| Age at time of report (years) | 13 | 5 | N/A | N/A | 19 | 9 | 7 | 8 |
| Age at time of diagnosis (years) | 13 | 2 | N/A | N/A | 19 | 9 | 7 | 2 |
| Gender | Female | Female | N/A | N/A | Male | Male | Female | Male |
| Variants | c.519T>A, p.(H173Q)/ c.731A>G, p.(Y244C) | c.G343G>A, p.(G115R)/ c.G343G>A, p.(G115R) | N/A | N/A | c.731A>G, p.(Y244C)/ c.605G>A, p.G202E | c.81A>C, p.(N27T)/ c.81A>C, p.(N27T) | c.81A>C, p.(N27T)/ c.81A>C, p.(N27T) | c.731A>G, p.(Y244C)/ c.536C>T, p.(P179L) |
| Inheritance | AR | AR | AR | AR | AR | AR | AR | AR |
| Ethnicity | Caucasian | Hispanic | N/A | N/A | Caucasian | N/A | N/A | Caucasian |
| Consanguinity | N/A | N/A | N/A | N/A | N/A | + | + | - |
| Failure to thrive/growth delay | + | + | N/A | N/A | + | + | - | + (transitory) |
| Neurodevelopmental findings | Mild DD, ID | DD | N/A | N/A | DD, moderate ID, OCD | Severe ID | Severe ID | DD/mild-moderate ID, autism spectrum disorder |
| Microcephaly | + | + | N/A | N/A | - | + | + | - |
| Structural brain abnormalities | - | - | N/A | N/A | Blake’s cyst Dysmorphic ventricles Cerebellar hypoplasia | Enlarged ventricles | Enlarged ventricles Corpus callosum dysgenesis Hypoplastic inferior vermis Enlarged cisterna magna | N/A |
| Ocular findings | Congenital cataracts | Congenital cataracts | N/A | N/A | Congenital cataract | Optic hypoplasia Strabismus Myopia Nystagmus | Optic hypoplasia Strabismus Myopia Nystagmus | Congenital cataracts Nystagmus |
| Skin findings (onset) | Generalized psoriasiform dermatitis (2 years) | Dry skin (N/A) | Psoriasiform dermatitis (early infancy) | Psoriasiform dermatitis (early infancy) | Mild scalp sebopsoriasis | Generalized psoriasiform dermatitis (2 months) | Perioral and anogenital psoriasiform dermatitis (3 years) | Transitory perianal psoriasiform dermatitis (7 years) |
| 4-monomethylsterol levels | ↑↑ | ↑↑* | ↑↑ | ↑↑ | ↑↑ | N/A | N/A | ↑↑ |
| 4,4′-dimethylsterol levels | ↑↑ | ↑↑* | ↑↑ | ↑↑ | ↑↑ | N/A | N/A | ↑↑ |
| Total cholesterol levels | ↓ | ↓ | N | N | ↓ | N | N | N |
| Therapy | Topical corticosteroids Calcipotriene Cyclosporine A Etanercept Phototherapy Oral isotretinoin | N/A | N/A | N/A | Atorvastatin (10 mg/d) Cholesterol (200 mg/TID) | Rosuvastatin (5 mg/d) Cholesterol (10 mg/kg/d) Topical vaseline + rosuvastatin 1% + cholesterol 10% | Rosuvastatin (5 mg/d) Cholesterol (10 mg/kg/d) Topical vaseline + rosuvastatin 1% + cholesterol 10% | Simvastatin (0.3 mg/kg/d) Pravastatin (4 mg/d) CoQ10 (50 mg/d) Topical cholesterol/simvastatin (transitory) |
| Miscellaneous findings | Delayed bone age Chronic arthralgia Delayed puberty | Joint contracture | Reported sibling of case 4 | Reported sibling of case 3 | Obesity Growth hormone deficiency Reduced NAA/Cr ratio (brain MRS) | ASD Unilateral cryptorchidism; Reported sibling of case 7 | Reported sibling of case 6 | - |
Our patient did not present with the characteristic SC4MOL deficiency clinical triad (MCCPD), but did exhibit congenital cataracts and developmental delay, both of which have improved with surgical and medical intervention, respectively. While growth delay does not appear to be a feature in our patient, there was early evidence of poor weight gain at 14 months of age (3rd centile). It is possible that the use of statin therapy in childhood has mitigated and prevented growth delay in our patient; however, he has yet to experience puberty and reach his adult height.
Psoriasiform dermatitis and congenital cataracts (or other ocular findings) may be considered characteristic findings in SC4MOL deficiency based on the higher frequency presented in this cohort [Table 1]. The presence of these features in an individual with neurodevelopmental impairment could raise the question of a cholesterol synthesis disorder. However, as other conditions have a similar presentation, some affected individuals were diagnosed only after an extensive work-up that eventually included plasma sterol analysis and MSMO1 sequencing to confirm the diagnosis [Table 1].
Of the variants identified in our proband, the paternally inherited likely pathogenic variant p.Y244C has been reported in trans in two non-consanguineous cases[7,10]. Interestingly, one of the other affected patients Case 5, [Table 1] carrying the p.Y244C variant is of Caucasian descent[10], particularly from Italian origin, a similar ethnic background to our patient based on the reported paternal ethnicity [Table 1]. In comparison, the p.P179L variant has not been reported in other affected individuals. However, its absence from healthy cohorts (NHLBI Exome Sequencing Project, 1000 Genomes Project), alteration of highly conserved amino acid, and deleterious predictions by in-silico models (Polyphen, SIFT), along with the biochemical evidence of SC4MOL dysfunction, support its pathogenicity.
Due to the enzyme’s proximal location and function in the post-squalene pathway[12], accumulation of methylsterols occurs[6,7]. This was evident in our patient, as well as five other reported individuals who also had elevated levels of methylsterols at diagnosis [Table 1]. Based on this deficiency, it is also expected that the distal end-product of this pathway (cholesterol) would show reduced concentrations. However, unlike the first case reported[7], our patient’s total cholesterol levels remained normal both before and after onset of therapy [Figure 2]. Furthermore, our review indicates that only 25% (n = 2) of affected individuals had low cholesterol levels at some point after diagnosis[6], suggesting the presence of residual enzymatic activity or possibly increased absorption of dietary cholesterol.
The molecular and biochemical evidence available is not sufficient to make detailed genotype-phenotype correlations or predictions of outcome at this point. Based on the limited number of cases reported, it appears that patients with homozygous variants tend to have a “classic” phenotype entailing psoriasiform dermatitis/dry skin, microcephaly, ocular findings and developmental delay/intellectual disability[6,11]. The variant p.N27T appears to have a severe presentation when homozygous, in comparison to the p.G115R variant[6,11]. The first case identified with the p.Y244C had a severe presentation compared to Case 5 and our patient [Table 1]. Therefore, individuals harboring the p.Y244C variant may vary in severity according to the second variant in trans.
The therapeutic goal of treating individuals with SC4MOL deficiency is to reduce methylsterol levels with statin therapy and provide the possibly deficient end-product, cholesterol, through dietary supplementation. Statins are used to decrease methylsterol and total cholesterol levels as they act by inhibiting HMG-CoA reductase, the third step in the pre-squalene pathway[13,14]. Inhibition of HMG-CoA reductase, in turn, results in downregulation of HMGCR, the gene that encodes this enzyme, and could potentially result in upregulation of genes involved in cholesterol synthesis, such as MSMO1, with residual SC4MOL activity resulting in a reduction of methylsterol levels[15,16]. Our patient was started on a simvastatin dose of 2 mg/day (0.25 mg/kg/d), and as he grew older, required 10 mg/day along with pravastatin. However, his methylsterol levels and methylsterol/total sterol ratio have continued to increase, which may indicate the need for further dose adjustment. Reported side effects of chronic statin administration include liver toxicity, myopathy, CK elevation, and possibly, but rarely, renal failure[13]. The latter is thought to be secondary to statin-induced myopathy (SAM)[17]. The etiology of SAM is unclear, but one possible mechanism could be mitochondrial dysfunction resulting from a reduction of ubiquinone (Coenzyme Q10)[17,18]. Our proband was monitored periodically for complications, and, aside from mild myalgias, no major side effects were exhibited. Continuation of CoQ10 therapy appeared to ameliorate this symptom.
Given that cholesterol is still required for multiple physiologic functions, supplementation or increased intake could be necessary. In the first patient described, a dose of 100 mg/kg/d was used in addition to statin therapy, leading to normalization of serum cholesterol levels and 20% reduction of methylsterols[6]. Two SC4MOL patients Cases 6 and 7, [Table 1], who had presented normal plasma cholesterol levels, were treated with statins and also supplemented with cholesterol[11]. However, because there is no clear data on the impact of cholesterol supplementation when the cholesterol level at diagnosis is normal, we chose not to include oral cholesterol supplementation or increased dietary intake in the treatment plan for our patient.
As evidenced by other conditions that present with similar skin findings, such as congenital hemidysplasia with ichthyosiform erythroderma and limb defects syndrome or Conradi-Hünermann-Happle syndrome, the use of a combined cholesterol/statin topical therapy has been beneficial[12]. It is thought that methylsterols affect cell cycles leading to skin cell hyperproliferation, causing a physiopathology reminiscent of psoriasiform dermatitis[7]. Therefore, the proposed mechanism of action for both oral statins and cholesterol also applies to topical administration. The cases affected by severe psoriatic dermatitis or related skin changes have shown a satisfactory response to combined cholesterol/statin topical therapy[6,11]. It is still not known why some SC4MOL patients do not exhibit severe skin related features. There could be multiple contributing factors such as residual enzyme function, for example, but this argument does not explain patients who presented with severe psoriatic dermatitis (cases 3, 4, 6 & 7) despite normal cholesterol levels. Our proband presented with intermittent perianal dermatitis, initially responding to traditional topical therapy. This eventually evolved into severe perianal psoriatic dermatitis that successfully responded to topical cholesterol/statin therapy without further recurrence.
In summary, SC4MOL deficiency is an ultra-rare condition with characteristic clinical features including microcephaly, cataracts, psoriatic dermatitis and neurodevelopmental problems. This disorder may be underdiagnosed given that some of these features overlap with other disorders and are relatively non-specific. In the presence of a mild case exhibiting only cataracts and developmental delay, sterol metabolism disorders, including SC4MOL deficiency, should be considered in the differential diagnosis, especially because of the availability of diagnostic testing and medical interventions. Further longitudinal data to assess the progression of this condition and the efficacy of therapy are needed.
DECLARATIONS
AcknowledgementWe would like to thank Richard I. Kelley, MD, PhD for the design and management of this patient’s statin therapy, the patient and his family, as well as the team of healthcare providers involved in this patient’s care.
Authors’ contributionsClinical care, conceptualization, methodology, writing of original draft, review and editing: Morales JA
Clinical care, supervision, review and editing: Curry CJ
Review and editing: Tise CG
Sterol measurements, resources, review and editing: Kratz L
Clinical care, supervision, review and editing: Enns GM
Availability of data and materialsThe data that support the findings of this study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationA written informed consent was reviewed and obtained from the proband’s parents.
Copyright© The Author(s) 2022.
REFERENCES
1. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol 2020;21:225-45.
2. Schade DS, Shey L, Eaton RP. Cholesterol review: a metabolically important molecule. Endocr Pract 2020;26:1514-23.
3. Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol 2000;16:459-81.
4. Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res 2011;52:6-34.
5. Herman GE, Kratz L. Disorders of sterol synthesis: beyond Smith-Lemli-Opitz syndrome. Am J Med Genet C Semin Med Genet 2012;160C:301-21.
6. He M, Smith LD, Chang R, Li X, Vockley J. The role of sterol-C4-methyl oxidase in epidermal biology. Biochim Biophys Acta 2014;1841:331-5.
7. He M, Kratz LE, Michel JJ, et al. Mutations in the human SC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis, microcephaly, and developmental delay. J Clin Invest 2011;121:976-84.
8. Byskov AG, Andersen CY, Nordholm L, et al. Chemical structure of sterols that activate oocyte meiosis. Nature 1995;374:559-62.
9. Xie H, Xia G, Byskov AG, Andersen CY, Bo S, Tao Y. Roles of gonadotropins and meiosis-activating sterols in meiotic resumption of cultured follicle-enclosed mouse oocytes. Mol Cell Endocrinol 2004;218:155-63.
10. Frisso G, Gelzo M, Procopio E, et al. A rare case of sterol-C4-methyl oxidase deficiency in a young Italian male: biochemical and molecular characterization. Mol Genet Metab 2017;121:329-35.
11. Kalay Yildizhan I, Gökpınar İli E, Onoufriadis A, et al. New homozygous missense MSMO1 mutation in two siblings with SC4MOL deficiency presenting with psoriasiform dermatitis. Cytogenet Genome Res 2020;160:523-30.
12. Elias PM, Crumrine D, Paller A, Rodriguez-Martin M, Williams ML. Pathogenesis of the cutaneous phenotype in inherited disorders of cholesterol metabolism: therapeutic implications for topical treatment of these disorders. Dermatoendocrinology 2011;3:100-6.
14. Kanungo S, Soares N, He M, Steiner RD. Sterol metabolism disorders and neurodevelopment-an update. Dev Disabil Res Rev 2013;17:197-210.
15. Zambrano T, Hirata RDC, Hirata MH, Cerda Á, Salazar LA. Statins differentially modulate microRNAs expression in peripheral cells of hyperlipidemic subjects: a pilot study. Eur J Pharm Sci 2018;117:55-61.
16. Mohammadzadeh N, Montecucco F, Carbone F, Xu S, Al-Rasadi K, Sahebkar A. Statins: epidrugs with effects on endothelial health? Eur J Clin Invest 2020;50:e13388.
17. Qu H, Guo M, Chai H, Wang WT, Gao ZY, Shi DZ. Effects of coenzyme Q10 on statin-induced myopathy: an updated meta-analysis of randomized controlled trials. J Am Heart Assoc 2018;7:e009835.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Morales JA, Curry CJ, Tise CG, Kratz L, Enns GM. Clinical characterization of a new individual with mild SC4MOL deficiency: diagnostic and therapeutic implications. J Transl Genet Genom 2022;6:257-65. http://dx.doi.org/10.20517/jtgg.2022.01
AMA Style
Morales JA, Curry CJ, Tise CG, Kratz L, Enns GM. Clinical characterization of a new individual with mild SC4MOL deficiency: diagnostic and therapeutic implications. Journal of Translational Genetics and Genomics. 2022; 6(2): 257-65. http://dx.doi.org/10.20517/jtgg.2022.01
Chicago/Turabian Style
Morales, J. Andres, Cynthia J. Curry, Christina G. Tise, Lisa Kratz, Gregory M. Enns. 2022. "Clinical characterization of a new individual with mild SC4MOL deficiency: diagnostic and therapeutic implications" Journal of Translational Genetics and Genomics. 6, no.2: 257-65. http://dx.doi.org/10.20517/jtgg.2022.01
ACS Style
Morales, JA.; Curry CJ.; Tise CG.; Kratz L.; Enns GM. Clinical characterization of a new individual with mild SC4MOL deficiency: diagnostic and therapeutic implications. J. Transl. Genet. Genom. 2022, 6, 257-65. http://dx.doi.org/10.20517/jtgg.2022.01
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.