
Estrogen and DNA damage modulate mRNA levels of genes involved in homologous recombination repair in estrogen-deprived cells

Abstract
Aims: Breast and ovarian cancers are frequently associated with mutations in genes involved in DNA double-strand break (DSB) repair by homologous recombination (HR). Risk factors for breast cancer are often linked to estrogen-related pathways. Here, we studied the crosslink between estrogen and the HR pathway.
Methods: We analyzed, using online annotation tolls, the enrichment of candidate estrogen-upregulated genes among DNA repair pathways. We analyzed how estrogen modulates mRNA levels of HR repair (HRR) genes in estrogen-receptor (ER)-positive cells. The cells were deprived of estrogen, and the mRNA levels of HRR genes were determined using real-time polymerase chain reaction, following estrogen addition as well as DNA damage induction. In addition, we examined the effect of estrogen on DNA repair, by immuno-fluorescence analysis, using the DSB marker phospho-histone H2AX, as an indicator for DSB repair. Finally, we performed a clonogenic survival assay to determine the effect of estrogen on cell survival.
Results: We discovered that genes whose mRNA levels are upregulated by estrogen are strongly associated with the HR pathway. We validated that estrogen upregulates mRNA levels of the HRR genes MRE11, RAD50, and PALB2, which have not been previously shown to be regulated by estrogen. Additionally, we revealed that DNA damage induces an upsurge in mRNAs encoding BRCA1, MRE11, RAD50, PALB2, and CtIP, in ER-positive cells deprived of estrogen. Notably, DSB repair was impaired in ER-positive cells deprived of estrogen, compared to cells exposed to the hormone. We also established that ER-positive cells deprived of estrogen are hypersensitive to DSBs.
Conclusion: These results suggest that exposure of ER-positive cells to estrogen triggers the expression of HRR genes, which is required to meet the increased repair demands due to the proliferating effect induced by estrogen. This may explain the higher chances of developing estrogen-dependent cancers due to mutations in HRR genes.
Keywords
INTRODUCTION
Genomic instability is a hallmark of cancerous cells, playing a crucial role in cancer initiation and progression. A main cause for genomic instability is DNA damage[1-5]. Of the various forms of DNA lesions, DNA double-strand breaks (DSBs) are considered highly toxic lesions which can trigger genomic instability[6]. DSBs occur constantly in cells by both endogenous and exogenous means[7,8]. The presence of DSBs in cells activates the DNA damage response (DDR), which senses and responds to the damage via extensive signaling networks. The DDR includes DNA repair, cell cycle checkpoint activation, cellular senescence, and apoptosis[8-12].
DSB repair occurs by two main mechanisms, the non-homologous end joining (NHEJ) and homologous recombination repair (HRR) pathways[7,8]. NHEJ involves direct ligation of broken ends, and it is considered error-prone since it often requires processing of the broken ends prior to ligation[13,14]. HRR uses a homologous sequence, most often the sister chromatid, as a template for DSB repair. Hence, HRR is restricted to the S and G2 phases of the cell cycle[13,15]. HRR is initiated by the resection of the 5' end, a process involving various proteins, including CtIP, the DSB sensor MRE11-RAD50-NBS1 (MRN) complex, BRCA1, BARD1, EXO1, DNA2, and BLM. The generated 3′ single-stranded DNA (ssDNA) tails are bound by RPA, which is later replaced by RAD51, a DNA-dependent ATPase that forms filaments on the DNA. The replacement of RPA with RAD51 requires BRCA2 and PALB2. RAD51 filaments function in searching for and invading to the homologous sequence, a process facilitated by RAD54. Next, RAD51 dissociates from the ssDNA, allowing base-pairing between the invading and complementary donor strands. This is followed by a strand extension, executed by DNA polymerase. The extended strand dissociates and anneals with the processed end of the non-invading strand on the opposite side of the DSB and invades to produce a double-Holliday junction that is resolved by BLM and additional helicases to yield crossover or non-crossover recombinants[13,15,16].
DNA repair pathways are frequently compromised in cancer cells. Particularly, faults in numerous HRR genes are associated with breast and ovarian cancers. Central to this are the hereditary breast cancer suppressors BRCA1 and BRCA2, which play a key role in HRR and are frequently mutated in familial breast and ovarian cancers. Other HR proteins, including PALB2, BLM, and the MRN complex, have also been associated with these cancers[17-20].
Estrogen is a group of sex steroid hormones that function in many physiological processes, including sexual and reproductive development in women. The predominant estrogen during a woman’s reproductive years is 17β-estradiol (E2)[21]. Estrogen executes its biological effects mainly by binding and activating two intracellular receptors, estrogen receptor (ER)α and ERβ, which are transcription factors. ERα activates pro-proliferative signals, targeting genes that promote cell proliferation or inhibit apoptosis. ERβ has anti-proliferative effects and thus antagonizes ERα[21-23]. Estrogen regulates the transcription of several HRR factors, such as BRCA1, BRCA2[24,25], and BLM[26]. There is a complex feedback system between BRCA1 and estrogen since BRCA1 also induces ERα mRNA expression as well as interacts with ERα and inhibits its mediated signaling[27-29].
The presence of estrogen in cells induces DNA damage[30-32]. Stimulation of estrogen results in a rapid increase in the formation of R-loops, which are co-transcriptional RNA-DNA products that are a source of DSBs[30]. Moreover, estrogen induces DSBs at the promotors of estrogen-inducible genes. These DSBs are specifically repaired by the HRR mechanism. Estrogen-induced DSB formation is dependent on ERα and the catalytic activity of topoisomerase IIβ[31]. Furthermore, estrogen metabolism leads to the accumulation of reactive oxygen species and nitric oxide moieties, resulting in oxidative DNA damage in the cells[32].
Previously, we conducted a bioinformatic screening aiming to find DDR genes that are upregulated by estrogen. This screening retrieved 1280 genes (a list of genes appears as Supplementary Table 1 in Ref.[33]). Here, we further analyzed these genes and determined that a high proportion of HRR genes are candidate estrogen-upregulated genes. We experimentally tested estrogen-dependent regulation of three HRR genes, MRE11, RAD50, and PALB2, which were indicated in the screening as candidate estrogen-upregulated genes but were not previously shown to be regulated by estrogen. We found that estrogen augments mRNA levels of these HRR genes in ER-positive breast cancer cells. Remarkably, the induction of DSBs in ER-positive cells deprived of estrogen results in increased levels of mRNA encoding BRCA1, MRE11, RAD50, PALB2, and CtIP. Moreover, we established that DSB repair is impaired in ER-positive cells deprived of estrogen and that these cells are hypersensitive to DNA damage. Collectively, our results indicate that estrogen triggers the expression of HRR genes simultaneously with its proliferative signal, in order to maintain genomic stability in the rapidly proliferating cells.
METHODS
Cell culture
MCF7 were cultured in Dulbecco’s modified Eagle’s medium (DMEM; MilliporeSigma, Massachusetts, USA) supplemented with 10% fetal bovine serum, 20 mM L-glutamine, 500 units/mL penicillin, and 0.5 mg/mL streptomycin (Biological Industries, Beit Haemek, Israel). E2 deprivation of MCF7 cells was preformed via culturing in DMEM lacking Phenol Red supplemented with 10% charcoal stripped fetal bovine serum, 20 mM L-glutamine, 500 units/mL penicillin, and 0.5 mg/mL streptomycin (Biological Industries, Beit Haemek, Israel). MCF10A cells were cultured in DMEM/F12 containing 5% horse serum, 20 ng/mL hEGF, 0.5mg/mL hydrocortisone, 10 ng/mL cholera toxin, 10 µg/mL human insulin (Millipore Sigma, Massachusetts, USA), 500 units/mL penicillin, and 0.5 mg/mL streptomycin (Biological Industries, Beit Haemek, Israel). E2 deprivation of MCF10A cells was performed by replacing horse serum with charcoal stripped fetal bovine serum. Cells were maintained in a humidified incubator at 37 °C with 5%
Quantitative real time PCR
Total RNA was extracted using EZ-RNA Isolation Kit reagent (Biological Industries, Beit HaEmek, Israel) from cells after a 72 h deprivation of E2, followed by reintroduction of 10 µM E2 for 3, 6, or 24 h, or the equivalent volume of EtOH for control. Additionally, cells were incubated with or without neocarzinostatin (NCS; 62.5 µg/mL final concentration in all cases, unless stated otherwise; MilliporeSigma, Massachusetts, USA) for 10 min; the NCS was then inactivated through a 10 min exposure to ambient light, and cells were left to recover in the incubator for 0, 1, or 6 h. The extracted RNA was reverse transcribed according to the manufacturer’s instructions of the High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, CA, USA). Quantitative real-time PCR was done using the StepOnePlus™ real-time PCR Systems (Applied Biosystems™, Thermofisher). The mRNA levels of each gene were normalized first to GAPDH mRNA levels under the same condition and then to the normalized value of the mRNA levels in E2-deprived cells for an estimation of expected fold change level. Primers were designed using the ROCHE Universal ProbeLibrary Assay Design Center software [Diagnostics GmbH Roche, Roche Life Science | Probes and The Universal ProbeLibrary, Roche Diagnostics GmbH (2009)]: BLM forward primer, TGTTCTGGCTGAGTGACGTT; BLM reverse primer, AGTTTGGATCCTGGTTCCGT; BRCA1 forward primer, ATCATTCACCCTTGGCACA; BRCA1 reverse primer, CATGGAAGCCATTGTCCTCT; MRE11 forward primer, AAGATGATGAAGTCCGTGAGG; MRE11 reverse primer, GAAGCAGACTCCTCTGACTGAGAT; PALB2 forward primer, CTTTCACGGCTCCATTTCAT; PALB2 reverse primer, AAAGGGCTCCACTGGTTTTT; RAD50 forward primer, AAACTGCGACTTGCTCCAGA; RAD50 reverse primer, GGCACAAGTCCCAGCATTTC; RAD51 forward primer, TCTCTGGCAGTGATGTCCTGGA; RAD51 reverse primer, TAAAGGGCGGTGGCACTGTCTA; GAPDH forward primer, TGCACCACCAACTGCTTAGC; and GAPDH reverse primer, GGCATGGACTGTGGTCATGAG.
Immunofluorescence
MCF7 cells were grown on coverslips. The cells were exposed to 10 µM E2, with the equivalent volume of EtOH for control, 62.5 µM NCS, or both and harvested after 0, 1, 4, 8, and 24 h of recovery. The cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100, then incubated for 2 h with mouse anti-phospho-histone H2A.X, Ser139 (γH2AX) antibody (MilliporeSigma, Massachusetts, USA), and washed three times in PBS and incubated for 1 h with Dylight 488 (Jackson Immunoresearch Laboratories, Pennsylvania, USA). After three washes, the nuclei were stained with 4′,6 diamidino-2-phenylindole (DAPI; MP Biomedicals, California, USA) and mounted on glass slides (DakoCytomation, Denmark). γH2AX foci were imaged by an Olympus IX81 fluorescence microscope with a 60 oil objective.
Clonogenic survival assay
MCF7 cells were seeded at densities of 4000 cells per 6 cm tissue culture dish along with 10 µM E2 or the equivalent volume of ethanol for control. The next day, cells were treated with varying doses of NCS (1, 2.5, 5, 7.5, and 10 ng/mL) and incubated for 12-14 days in Ham’s F-10 nutrient mixture medium supplemented with 20% fetal bovine serum (Biological Industries, Beit Haemek, Israel), 20 mM L-glutamine, 500 units/mL penicillin, and 0.5 mg/mL streptomycin (Biological Industries, Beit Haemek, Israel). Cell colonies were fixed and stained with 2% (w/v) crystal violet in 50% ethanol, as in Ref.[34]. Colonies of at least 50 cells were scored. The surviving fraction for each NCS dose was calculated and survival curves were constructed.
Statistical analysis
Statistical analyses were conducted using the Kruskal-Wallis one-way analysis of variance test, using the Prism software package (8th edition, GraphPad, California, United States).
RESULTS
Many estrogen-upregulated genes are linked to the HR pathway
Hereditary breast and ovarian cancers are often associated with mutations in DNA repair genes, most of which play a direct role in HRR[35]. Since these tissues are estrogen-responsive, it implies a link between estrogen and the HRR pathway. Previously, we conducted a bioinformatic screening aimed at identifying genes that are upregulated by estrogen[33]. Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of the predicted 1280 estrogen-upregulated genes retrieved from the screening indicated an enrichment for the HR pathway [Figure 1A]. We further examined the connection between the HR pathway and estrogen by determining the percentage of the predicted estrogen-upregulated genes among the genes that play a role in the pathway. The list of genes that play a role in the HR pathway was obtained using the AmiGO annotation tool[36]. Overall, 31% of the established HRR genes were retrieved as candidate estrogen-upregulated genes in the screening [Figure 1B]. When analyzing proportions of the candidate estrogen-upregulated genes among other repair pathways, we found that lower percentages of candidate estrogen-upregulated genes (24.7%, 20.9%, 20.5%, and 10.4%) are annotated in NHEJ, base excision repair (BER), mismatch repair (MMR), and nucleotide excision repair (NER), respectively [Figure 1B]. Taken together, these results confirm that a large fraction of DNA repair genes, and of the HR pathway genes in particular, are regulated by estrogen.
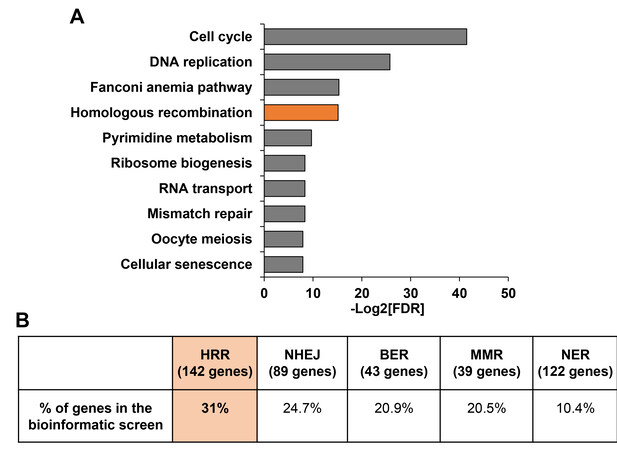
Figure 1. The HR pathway is enriched for candidate estrogen-upregulated genes. (A) The top ten significantly enriched pathways among candidate estrogen-upregulated genes. (B) Summary of the proportion of candidate estrogen-upregulated genes among the genes that play a role in numerous DNA repair pathways, according to AmiGO. FDR: False discovery rate; HRR: homologous recombination repair; NHEJ: non-homologous end joining; BER: base excision repair; MMR: mismatch repair; NER: nucleotide excision repair.
We studied the extent of estrogen involvement in the HR pathway by examining whether 13 genes which play central roles in HRR were retrieved in the screening as candidate estrogen-upregulation genes (BARD1, BLM, BRCA1, BRCA2, CtIP, DNA2, EXO1, MRE11, NBN, PALB2, RAD50, RAD51, and RAD54). All of these HRR genes, apart from CtIP, were retrieved from the bioinformatic screening as genes upregulated by estrogen[33]. Seven out of these genes, namely BARD1[37], BLM[26], BRCA1, BRCA2[24,25], DNA2[33], RAD51[38], and RAD54[33], are known to be regulated by estrogen. Of note, DNA2 and RAD54 were previously validated in our lab as upregulated by estrogen[33]. CtIP was not identified in the screening, but its connection to estrogen was published elsewhere[39]. Five of the 13 key HRR genes, namely EXO1, NBN, MRE11, RAD50, and PALB2, were identified only in the bioinformatic screening as candidate estrogen-upregulated genes.
Estrogen augments mRNA levels of MRE11, RAD50, and PALB2 genes
To determine whether estrogen regulation is a hallmark of HRR genes, we examined estrogen regulation of three genes, MRE11, RAD50, and PALB2, which were identified in the bioinformatic screening as candidate estrogen-upregulated genes. Since CtIP appeared as an estrogen-upregulated gene in the literature[39], but not in the bioinformatic screening[33], we also analyzed CtIP regulation by estrogen. As a positive control, we tested BLM, whose regulation by estrogen is well documented[26]. We studied changes in mRNA levels of those HRR genes between ER-positive MCF7 breast carcinoma cells that were deprived of E2 for 72 h and then resupplied with the hormone, and cells that were resupplied with EtOH, as a control. The real-time PCR results indicate that mRNA levels of MRE11, RAD50, PALB2, and CtIP, as well as of the positive control, BLM, were significantly elevated following reintroduction of E2 to the cells [Figure 2A]. The augmentation, due to re-supplementation of E2, was observed from the first time point analyzed (3 h after E2 addition), peaked at the 6 h time point, and remained significantly higher 24 h after the cells were re-supplemented with E2 [Figure 2A].
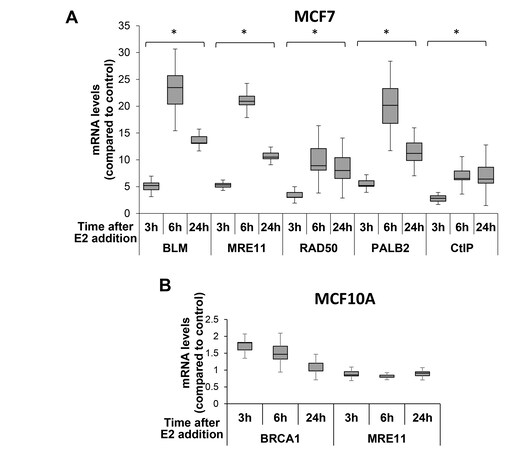
Figure 2. mRNA levels of HRR genes augments in the presence of E2 in ER-positive cells. MCF7 (A) or MCF10A (B) cells were deprived of E2 for 72 h. Next, E2 was added to the media for the indicated time points. mRNA was extracted, and quantitative real-time PCR was performed. mRNA levels of HRR genes were normalized to GAPDH and compared to the control. Notice changes in y-axis scale between (A) and (B). Data of four experiments, for each cell line, are shown. *A significant change compared to control; P-value < 0.05. HRR: Homologous recombination repair.
Following the finding that estrogen resubmission augments mRNA levels of HRR genes, we confirmed that this effect is unique to cells that express ERs. E2 was reintroduced to ER-negative MCF10A epithelial breast cells[40] that were deprived of estrogen for 72 h. As a positive control, we tested BRCA1 that is an established estrogen-upregulated gene[24,25]. There was no significant change in mRNA levels of MRE11 as well of the control BRCA1, due to E2 addition in MCF10A cells [Figure 2B].
mRNAs encoding HRR proteins are augmented upon DNA damage induction in ER-positive cells deprived of estrogen
We discovered that mRNA levels of MRE11, RAD50, and PALB2 increase upon introduction of estrogen to ER-positive cells [Figure 2A]. This is similar to addition HRR genes[24-26,33,37-39]. Furthermore, estrogen induces DSBs in these cells[30-32]. We next determined the crosstalk between estrogen regulation of HRR genes and DNA damage induction. When we cultured MCF7 cells in growth media containing E2 and induced DNA damage, with the radiomimetic drug NCS, none of the tested HRR genes showed a significant change in mRNA levels at all-time points analyzed after DNA damage induction [Figure 1A]. This is in line with the regulation of DSB repair by post-translational modifications[11,41-44]. Next, we analyzed mRNA levels of BRCA1, MRE11, RAD50. PALB2, and CtIP in E2-deprived MCF7 cells that were exposed to NCS and were either harvested immediately after inactivation of the DNA damaging agent (0 h time point) or left to recover for 1 or 6 h after DNA damage induction. Notably, induction of DSBs in E2-deprived MCF7 cells resulted in an augmentation of mRNA levels of all analyzed HRR genes at all examined time points [Figure 3A]. The rise in mRNAs encoding HRR proteins occurred immediately after NCS inactivation (0 h time point; [Figure 3A]). The maximum augmentation was obtained 1 h after damage induction for BLM, MRE11, RAD50, and PALB2, while CtIP mRNA levels peaked after 6 h [Figure 3A]. Moreover, the extent of the augmentations of mRNA levels of most analyzed HRR genes that was obtained when either E2 or NCS were added to E2-deprived MCF7 cells was at similar magnitudes. For RAD50 and CtIP, mRNA augmentation was higher due to DNA damage induction (compare [Figures 2A and 3A]).
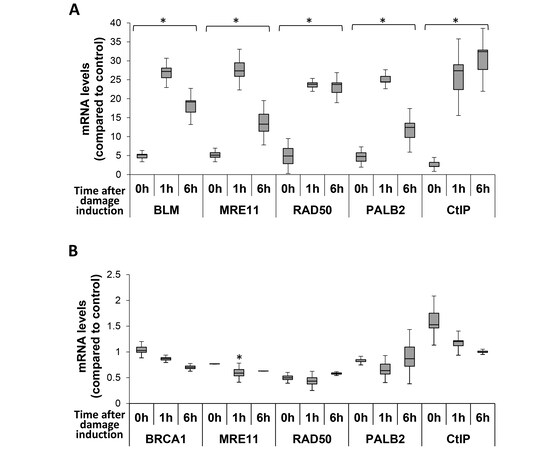
Figure 3. mRNA levels of HRR genes augmented after the induction of DNA damage in ER-positive cells deprived of estrogen. MCF7 cells were grown in the absence of E2 for 72 h. Next, DSBs were induced with NCS for 10 min following by NCS inactivation (A) or E2 was supplemented for 24 h followed by DSB induction (B). Cells were allowed to recover from DSB induction for the indicated time points, mRNA was extracted, and quantitative real-time PCR was performed. mRNA levels of HRR genes were normalized to GAPDH and compared to the control. Notice changes in Y-axis scale between (A) and (B). The results from four experiments are shown. *A significant change compared to control; P-value < 0.05. DSB: Double-strand break.
Next, we verified that the increase in mRNA levels of HRR genes due to the induction of DNA damage in E2-deprived MCF7 cells is due to lack of E2. MCF7 cells were grown without E2 for 72 h, after which E2 was reintroduced for 24 h. Next, DSBs were induced in the cells with NCS. mRNA levels of HRR genes were determined at the indicated time points. mRNA levels of BRCA1, MRE11, RAD50, PALB2 and CtIP were not augmented following DSB induction [Figure 3B], indicating that the augmentation in mRNA levels of mRNA due to DSB formation occurs only in E2-depleted cells.
Next, to validate that the elevation in mRNA levels of HRR genes due to the induction of DNA damage is specific to cells that respond to estrogen, we analyzed the effect of DSB formation on MCF10A ER-negative cells. E2-deprived MCF10A cells were grown for 72 h followed by exposure to NCS for DSB induction. The cells were collected immediately (0h) or left to recover for 1 or 6 h. Notably, mRNA levels of BRCA1 and MRE11 showed no significant rise, but rather a decrease in quantity immediately after (0 h) and following 1 h of NCS treatment [Figure 1B]. Of note, the reduction of mRNA levels of the analyzed HRR genes in ER-negative cells is an order of magnitude lower compared to the increase in the mRNA levels of the analyzed genes in ER-positive cells (compare [Figure 1B] and [Figure 3A]). Further experiments should be done to reveal the effect of estrogen on ER-negative cells. Collectively, our results show that ER-positive cells deprived of estrogen react rapidly to the presence of DNA damage and augment mRNA levels of HRR genes. This effect is unique to cells grown in an environment lacking estrogen, as cells that were constantly grown with E2 did not show changes in HRR gene mRNA levels due to damage induction.
DSB repair is impaired in ER-positive cells deprived of estrogen
We found that estrogen, as well as DNA damage induction, increases the amount of mRNAs encoding HRR proteins in ER-positive cells that were grown without estrogen [Figures 2 and 3]. This suggests that estrogen affects DSB repair in ER-positive cells. To study the effect of estrogen on DSB repair, we initially analyzed DSB repair in E2-deprived MCF7 cells. MCF7 cells were deprived of E2 for 72 h and then DSBs were induced with NCS. The cells were harvested immediately following NCS inactivation (0 h) or left to recover for 1, 4, 8, or 24 h before fixation. The fixated cells were subjected to Immunofluorescence (IF) analysis, with antibodies directed against the phosphorylated form of histone H2AX (γH2AX). γH2AX level reflects the amount of DSBs in cells. The formation of γH2AX foci is an indication for DSB appearance and signaling (seen at early time points after DSB induction), and their disappearance indicates DNA repair (seen at later time points)[45,46]. As expected, NCS induced γH2AX focus formation [Figure 4A] (DNA damage panels) and [Figure 4B]. Augmentation in the amount of γH2AX foci in the cells was observed from the time of DNA damage induction (0 h) until 8 h after DNA damage induction. The amount of DSBs, as reflected by γH2AX focus formation, was highest immediately and 1 h after DNA damage induction, and from the 4 h time point there was a reduction in γH2AX focus formation, indicating that the DSB repair process had begun. Then, 24 h after DNA damage induction, the amount of γH2AX foci was similar to the control, untreated cells, indicating that the repair process was successfully completed [Figure 4B]. Next, we studied whether estrogen modulated DSB repair in these cells. E2-deprived MCF7 cells were supplemented with E2. Twenty minutes after E2 addition, DSBs were induced with NCS. γH2AX focus formation was examined at different time points after DSB induction. γH2AX foci appeared rapidly after DNA damage induction and stayed 1 h post damage induction ([Figure 4A] (E2 + DNA damage panels) and [Figure 4C]. DSB repair occurred 4 h after DNA damage induction, as reflected by the return of the amount of γH2AX foci to that of the control, untreated cells, at this time point [Figure 4C]. Markedly, DSB repair took longer in cells deprived of estrogen compared to cells that were re-exposed to the hormone (24 h compared to 4 h; compare in
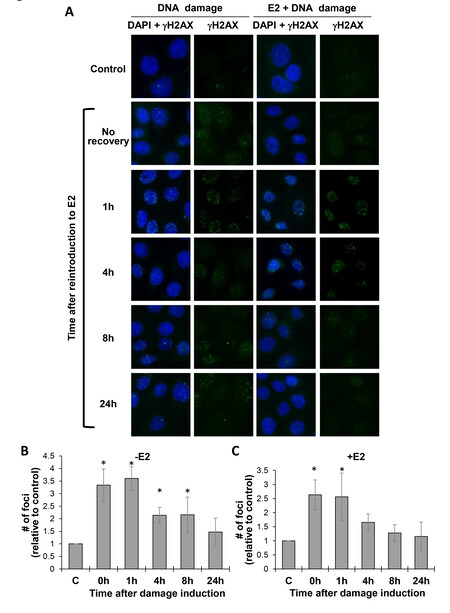
Figure 4. DSB repair is impaired in ER-positive cells-deprived from estrogen. MCF7 cells were deprived of E2 for 72 h and either exposed to NCS for 10 min (DNA damage) (A,B) or resupplied with E2 and after 20 min were exposed to NCS for 10 min (E2 + DNA damage) (A,C). NCS was inactivated and cells were left to recover for the indicated time points and IF analyses, with anti γH2AX-antibody and DAPI, were performed. (A) Representative images. (B,C) Quantification of the average number of foci per cell at each time point following DSB induction. - E2 - DNA damage without E2 addition; + E2 - DNA damage and E2 addition. At least 50 cells were analyzed for each time point. The results from four experiments are shown. *A significant change compared to control (C); P-value < 0.05. DSB: Double-strand break; NCS: Neocarzinostatin.
ER-positive cells deprived of estrogen are hypersensitive to DNA damage
We discovered that ER-positive cells deprived of E2 possess a lower capacity for DNA repair [Figure 4]. Next, we determined the effect of E2 deprivation on cell survival. We performed a clonogenic survival assay on MCF7 cells that were deprived of estrogen for 72 h and then supplemented with either E2 or EtOH (control) for 24 h. Then, DNA damage was induced by exposing the cells to growing amounts of NCS. After 10 days in culture, cell colonies were fixed and stained, and colonies consisting of more than 50 cells were counted. As expected, the number of colonies obtained from cells that were grown in the presence of E2 was 1.71-fold higher compared to the number of colonies obtained from cells grown without E2 [Figure 5A]. This supports the proliferating effect of estrogen. Next, we normalized the amount of colonies obtained at each DNA damage dose to that of its control condition (E2-deprived or -supplemented cells without DNA damage). The comparison of the ratio obtained for cells deprived of E2 and that of cells exposed to the hormone revealed that, in the presence of DNA damage, more cells survive when supplemented with E2 [Figure 5B]. These results are in line with impairment of DSB repair in cells starved for estrogen compared with cells supplemented with the hormone [Figure 4].
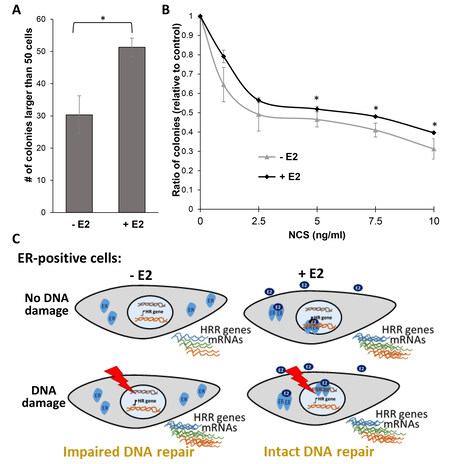
Figure 5. Deprivation of estrogen sensitizes ER-positive cells to DNA damage. (A, B) MCF7 cells were deprived of E2 for 72 h. E2 (+ E2) or EtOH (- E2) was added to cells for 24 h, and then the indicated amounts of the DNA-damaging agent Neocarzinostatin (NCS) were added. Ten days after seeding, the cells were stained with crystal violet and colonies of ≥ 50 cells were counted. (A) Average number of colonies obtained from cells not treated with NCS. (B) Clonogenic survival assay, analyzing the ratio of colonies compared with treatment control. The results from three experiments are shown. *A significant change compared to control; P-value < 0.05. (C) A model depicting the interplay among estrogen, HRR factors, and DNA damage in ER-positive cells. In E2-deprived cells, there is a basal level of mRNA encoding HRR factors. Upon supplementation of E2, the level of these mRNAs is elevated. Induction of DNA damage in E2-cells deprived cells also results in augmentation of mRNA levels of HRR genes. On the contrary, induction of DNA damage in cells that were grown in the presence of E2 did not result in increased levels of HRR genes mRNAs. HRR: Homologous recombination repair.
DISCUSSION
We showed that estrogen is a master regulator of HRR. Estrogen has previously been shown to be a regulator of several HRR genes, such as BRCA1, BRCA2, and BLM[24-26]. Here, we demonstrated that estrogen also regulates the expression of MRE11, RAD50, and PALB2. We also showed that genes that were identified as upregulated by estrogen in a bioinformatic screening[33] are enriched for the HRR pathway. Furthermore, we found a high proportion of estrogen-upregulated genes among genes that play a role in the HRR pathway. Our results support the following model [Figure 5C]: In ER-positive cells deprived of estrogen, there is a basal level of mRNAs encoding HRR proteins. Supplementing estrogen, as well as introducing DNA damage to these cells, results in an upsurge of mRNAs of HRR genes. Estrogen increases the mRNA levels of HRR genes, probably to facilitate DNA damage repair. DNA damage is a byproduct of estrogen metabolism as well as of proliferation, which is triggered by estrogen[21-23,30-32]. Furthermore, DSB formation in ER-positive cells deprived of estrogen results in an increment of mRNAs encoding HRR proteins [Figure 5C]. This is probably because these cells are more sensitive to DNA damage and augmentation of repair factors may compensate this sensitivity. However, the rise in the levels of mRNAs encoding HRR proteins will not provide an immediate protection from DNA lesions induced by estrogen, as it takes time for a translation to take place[47-49]. This is in correlation to our finding that DSB repair in E2-deficient cells is less efficient than in E2-supplemented cells [Figure 5C]. In correlation with impaired DSB repair in ER-positive cells deprived of estrogen, these cells are hypersensitive to DSB formation. Breast cancer is the most common cancer among women in the western world, with a one in eight chance for women to develop breast cancer during their lifetime. Risk factors for breast cancer are mainly associated with estrogen-related pathways. An increased exposure to estrogen due to early menarche, late menopause, and absence of childbearing amplify the risk of breast cancer. Furthermore, aberrant hormone exposure, whether induced naturally or via clinical administration, increase the risk of breast cancer[50-53]. Here, we disclosed a crosstalk between estrogen and DNA repair. We found that ER-positive cells are sensitive to both estrogen and DNA damage and that cells deprived of estrogen have impaired DSB repair. The addition of estrogen induces DNA damage in these cells and triggers genomic instability[30-32], which is a hallmark of cancer cells[1-5]. Consequently, changes in estrogen levels in the female body will have an impact on ER-positive cells, which may trigger genomic instability. Early menarche, late menopause, and menopausal hormone therapy prolong estrogen presence in the female body and may induce genomic instability to ER-positive cells, as indicated by our results that demonstrate a crucial role of estrogen in DNA repair. This may explain, at least in part, the etiology of estrogen-related cancers. Women with hereditary breast and ovarian cancer syndrome have an increased lifetime risk for developing breast and ovarian cancers as well as other cancer types. They are diagnosed at a younger age, compared to women with sporadic breast or ovarian cancers. While mutations in BRCA1 and BRCA2 genes are responsible for the majority of these cancers, other cancer susceptibility genes exist. Among these are numerous genes that play a role in HRR, such as PALB2, RAD51C/D, XRCC2, MRE11, RAD50, and NBN[54,55]. Estrogen regulates transcription of several of these susceptibility genes, such as BRCA1 and BRCA2[24,25]. Here, we found that all susceptibility genes listed above were retrieved in a bioinformatic screening for genes upregulated by estrogen[33]. Furthermore, we validated that estrogen regulates the expression of the susceptibility breast and ovarian cancer genes PALB2, MRE11, and RAD50. Thus, regulation by estrogen is a general feature of HRR genes. Moreover, we validated that two members of the MRN complex (MRE11 and RAD50) are upregulated by estrogen. Since NBN was retrieved in the bioinformatic screening as a candidate estrogen-upregulated gene, we speculate that estrogen modulates the levels, and thus the activity, of the MRN complex, which plays key roles not only in HRR but also in DNA damage recognition and signaling[56]. We further propose that the HRR pathway is specifically essential for estrogen-dependent cells. In these cells, the quantity of mRNAs encoding HRR proteins is tightly regulated. Cases of estrogen deprivation, exposure to the hormone or DNA damage induction, result in augmentation of mRNA levels of HRR genes. In cases of hereditary mutation in a HRR gene, fine-tuning of the amounts of HRR mRNAs is impaired, resulting in the misregulation of the HRR pathway. Defects in HRR, as in other DNA repair pathways, result in accumulation of mutations and chromosomal defects, leading to genomic instability, which drives cancer development[5]. Interestingly, we found that genes that take part in DNA repair pathways, such as NHEJ, BER, MMR, and NER, are enriched for estrogen-upregulated HRR genes. This enrichment was less profound compared to HRR. ER regulates all these repair pathways. For example, ER directly binds members of BER (e.g., FEN1 and APE1), NHEJ (Ku70 and Ku86), and MMR (MSH2). These interactions either enhance repair activity or modulate ER activity. In addition, estrogen addition affects NER efficiency[22]. However, predisposition to breast and ovarian cancers results mainly from mutations in HRR genes, suggesting that HRR pathway crosstalk with estrogen is unique.
We demonstrated that DNA damage induction in ER-positive cells grown in the presence of estrogen does not result in augmentation of mRNAs encoding HRR proteins, while in estrogen-deprived cells it results in such augmentation. In correlation, we found that estrogen-supplemented cells cope better with DNA damage induction, compared to cells deprived of estrogen. Gene expression includes four steps: transcription, mRNA degradation, translation, and protein degradation. Each step is a highly controlled process that takes some time. For example, the rate of ribosome translocation on a single mRNA molecule is 3–5 codons per second[47-49]. Therefore, the increased mRNA levels of HRR factors due to DNA damage induction is not expected to result in an immediate impact on DNA repair. Notably, Li et al. demonstrated a higher HRR frequency in MCF7 cells deprived of E2 compared to cells re-supplemented with E2[38]. This is despite the addition of E2 to MCF7 cells resulting in proliferation and cell accumulation in the G2/M phase[21-23,57], and HRR is known to occur during S and G2 phases of the cell cycle[13,15]. In their study, Li et al. used the highly studied DR-GFP reporter cassette for evaluating HRR[38]. Since the nuclease is present in cells for a prolonged time, and NHEJ can be accurate in this restriction enzyme system, there may be several cycles of DSB formation and repair by precise NHEJ. Moreover, there is a delay between transfection of the I-SceI-expressing plasmid and the time at which the endonuclease enters the nucleus and starts to induce DSBs[58-60]. Our results may explain the higher HRR efficiency obtained in cells deprived of estrogen, when using the DR-GFP system[38]; DSB induction in cells deprived of estrogens results in an immediate rise in mRNAs encoding HRR factors. These transcripts are translated to HRR factors throughout the process of cycles of DSB formation by I-SceI and repair by precise NHEJ, eventually resulting in more efficient HRR repair. It would be interesting to compare the adaptation to DNA damage induction in cells deprived of estrogen and cells resupplied with the hormone.
Estrogen mediates its physiological functions mainly via ERα and ERβ. The binding of estrogen to these receptors triggers their dimerization and translocation into the nucleus. This results in a direct regulation of gene expression[61,62]. Apart from this ER-dependent genomic regulation, estrogen also mediates cellular activities by a non-genomic pathway through binding to G protein-coupled ER (GPER). It was demonstrated that estrogen induces, via GPER, proliferation in MCF10A cells[63]. GPER is highly expressed in most breast cancer cells and high GPER expression is strongly correlated with a poor prognosis in both breast and ovarian cancers. In ER-positive cells, estrogen, apart from binding to the ERs, may activate GPER as a parallel or alternative pathway. In ER-negative cells, estrogen effect via GPER will be the dominant pathway. This suggests that estrogen deprivation will be sensed not only by ER-positive cells but also by ER-negative cells, through the GPER pathway[64]. Indeed, we showed here that induction of DNA damage to ER-negative MCF10A cells deprived of estrogen results in a slight reduction in mRNA levels of HRR genes. Further studies aim at elucidating the roles of ERs and GPER in HRR genes regulation will determine the interplay among DNA repair, these receptors, and estrogen.
In conclusions, this study revealed a strong correlation between estrogen and DSB repair. We showed that estrogen is a master regulator of the levels of mRNAs of HRR genes. We also found that DSB formation in ER-positive cells deprived of estrogen results in augmentation of mRNAs encoding proteins involved in HRR. Estrogen is required for proper DSB repair, and cells deprived of estrogen are hypersensitive to DNA damage. The effect of estrogen on DSB repair clarifies the connection between the hormone and estrogen-related cancers.
DECLARATIONS
AcknowledgmentsWe thank members of the Goldberg and Kerem labs for assistance with this study. Specifically, to Ruth Viner-Breuer, Merav Gold and Amir Hadad for critical reading of this manuscript, and Dan Sarni and Eyal Itskovits for advice regarding the statistical analysis.
Authors’ contributionsMajor contribution to the conception and methodology of the study, funded the study and supervised and validated it, wrote the manuscript: Goldberg M
Made contributions to conception and design of the study and performed experiments, analysis and interpretation: Zach L
Contributed to the validation of the study, perform experiments and was involved in interpretation of data for the work: Yedidia Aryeh L
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Israel Science Foundation, grant number 684/13.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Ferguson LR, Chen H, Collins AR, et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol 2015;35 Suppl:S5-S24.
2. Bartkova J, Horejsí Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005;434:864-70.
5. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability-an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010;11:220-8.
6. Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol 2013;20:648-59.
7. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012;47:497-510.
8. Pardo B, Gómez-González B, Aguilera A. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell Mol Life Sci 2009;66:1039-56.
9. Riches LC, Lynch AM, Gooderham NJ. Early events in the mammalian response to DNA double-strand breaks. Mutagenesis 2008;23:331-9.
11. Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007;19:238-45.
12. Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. Paving the way for γH2AX phosphorylation: chromatin changes in the DNA damage response. Cell Cycle 2009;8:1494-500.
13. Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 2019;20:698-714.
14. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 2010;79:181-211.
15. Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 2013;5:a012740.
16. Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res 2008;18:134-47.
17. Trenner A, Sartori AA. Harnessing DNA double-strand break repair for cancer treatment. Front Oncol 2019;9:1388.
18. Bogdanova N, Helbig S, Dörk T. Hereditary breast cancer: ever more pieces to the polygenic puzzle. Hered Cancer Clin Pract 2013;11:12.
19. Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 2015;7:a016600.
20. Hsu HM, Wang HC, Chen ST, Hsu GC, Shen CY, Yu JC. Breast cancer risk is associated with the genes encoding the DNA double-strand break repair Mre11/Rad50/Nbs1 complex. Cancer Epidemiol Biomarkers Prev 2007;16:2024-32.
22. Caldon CE. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front Oncol 2014;4:106.
23. Nilsson S, Mäkelä S, Treuter E, et al. Mechanisms of estrogen action. Physiol Rev 2001;81:1535-65.
24. Spillman, A.M. Bowcock. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene 1996;13:1639-45.
25. Cabanes A, Wang M, Olivo S, et al. Prepubertal estradiol and genistein exposures up-regulate BRCA1 mRNA and reduce mammary tumorigenesis. Carcinogenesis 2004;25:741-8.
26. Iso T, Futami K, Iwamoto T, Furuichi Y. Modulation of the expression of bloom helicase by estrogenic agents. Biol Pharm Bull 2007;30:266-71.
27. Marks JR, Huper G, Vaughn JP, et al. BRCA1 expression is not directly responsive to estrogen. Oncogene 1997;14:115-21.
28. Gorski JJ, Kennedy RD, Hosey AM, Harkin DP. The complex relationship between BRCA1 and ERalpha in hereditary breast cancer. Clin Cancer Res 2009;15:1514-8.
29. Wang L, Di LJ. BRCA1 and estrogen/estrogen receptor in breast cancer: where they interact? Int J Biol Sci 2014;10:566-75.
30. Stork CT, Bocek M, Crossley MP, et al. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 2016;5:e17548.
31. Williamson LM, Lees-Miller SP. Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 2011;32:279-85.
32. Rajan A, Nadhan R, Latha NR, Krishnan N, Warrier AV, Srinivas P. Deregulated estrogen receptor signaling and DNA damage response in breast tumorigenesis. Biochim Biophys Acta Rev Cancer 2021;1875:188482.
33. Strauss C, Kornowski M, Benvenisty A, et al. The DNA2 nuclease/helicase is an estrogen-dependent gene mutated in breast and ovarian cancers. Oncotarget 2014;5:9396-409.
34. Baranes-Bachar K, Levy-Barda A, Oehler J, et al. The Ubiquitin E3/E4 Ligase UBE4A Adjusts Protein Ubiquitylation and Accumulation at Sites of DNA Damage, Facilitating Double-Strand Break Repair. Mol Cell 2018;69:866-878.e7.
35. Kleibl Z, Kristensen VN. Women at high risk of breast cancer: molecular characteristics, clinical presentation and management. Breast 2016;28:136-44.
36. Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S. AmiGO Hub. AmiGO: online access to ontology and annotation data. Bioinformatics 2009;25:288-9.
37. Creekmore AL, Ziegler YS, Bonéy JL, Nardulli AM. Estrogen receptor alpha regulates expression of the breast cancer 1 associated ring domain 1 (BARD1) gene through intronic DNA sequence. Mol Cell Endocrinol 2007;267:106-15.
38. Li Z, Chen K, Jiao X, et al. Cyclin D1 integrates estrogen-mediated DNA damage repair signaling. Cancer Res 2014;74:3959-70.
39. Inoue A, Yoshida N, Omoto Y, et al. Development of cDNA microarray for expression profiling of estrogen-responsive genes. J Mol Endocrinol 2002;29:175-92.
40. Qu Y, Han B, Yu Y, et al. Evaluation of MCF10A as a reliable model for normal human mammary epithelial cells. PLoS One 2015;10:e0131285.
41. Garvin AJ. Beyond reversal: ubiquitin and ubiquitin-like proteases and the orchestration of the DNA double strand break repair response. Biochem Soc Trans 2019;47:1881-93.
42. Oberle C, Blattner C. Regulation of the DNA damage response to DSBs by post-translational modifications. Curr Genomics 2010;11:184-98.
43. Her J, Bunting SF. How cells ensure correct repair of DNA double-strand breaks. J Biol Chem 2018;293:10502-11.
44. Dantuma NP, van Attikum H. Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J 2016;35:6-23.
45. Mah LJ, El-Osta A, Karagiannis TC. GammaγH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 2010;24:679-86.
46. Sharma A, Singh K, Almasan A. Histone γH2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 2012;920:613-26.
47. Schwanhäusser B, Busse D, Li N, et al. Global quantification of mammalian gene expression control. Nature 2011;473:337-42.
48. Yan X, Hoek TA, Vale RD, Tanenbaum ME. Dynamics of translation of single mRNA molecules in vivo. Cell 2016;165:976-89.
49. Wang C, Han B, Zhou R, Zhuang X. Real-time imaging of translation on single mRNA transcripts in live cells. Cell 2016;165:990-1001.
50. Dall GV, Britt KL. Estrogen effects on the mammary gland in early and late life and breast cancer risk. Front Oncol 2017;7:110.
51. Group on Hormonal Factors in Breast Cancer. Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence. Lancet 2019;394:1159-68.
52. Wang K, Li F, Chen L, Lai YM, Zhang X, Li HY. Change in risk of breast cancer after receiving hormone replacement therapy by considering effect-modifiers: a systematic review and dose-response meta-analysis of prospective studies. Oncotarget 2017;8:81109-24.
53. Lyytinen H, Pukkala E, Ylikorkala O. Breast cancer risk in postmenopausal women using estrogen-only therapy. Obstet Gynecol 2006;108:1354-60.
54. Apostolou P, Fostira F. Hereditary breast cancer: the era of new susceptibility genes. Biomed Res Int 2013;2013:747318.
55. Reginato G, Cejka P. The MRE11 complex: a versatile toolkit for the repair of broken DNA. DNA Repair (Amst) 2020;91-2:102869.
56. Walsh CS. Two decades beyond BRCA1/2: homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol Oncol 2015;137:343-50.
57. Tian JM, Ran B, Zhang CL, Yan DM, Li XH. Estrogen and progesterone promote breast cancer cell proliferation by inducing cyclin G1 expression. Braz J Med Biol Res 2018;51:1-7.
58. Brandsma I, Gent DC. Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr 2012;3:9.
59. Shahar OD, Raghu Ram EV, Shimshoni E, Hareli S, Meshorer E, Goldberg M. Live imaging of induced and controlled DNA double-strand break formation reveals extremely low repair by homologous recombination in human cells. Oncogene 2012;31:3495-504.
60. Nagy Z, Soutoglou E. DNA repair: easy to visualize, difficult to elucidate. Trends Cell Biol 2009;19:617-29.
61. Jia M, Dahlman-Wright K, Gustafsson JÅ. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab 2015;29:557-68.
62. Bolton JL, Thatcher GR. Potential mechanisms of estrogen Quinone carcinogenesis. Chem Res Toxicol 2008;21:93-101.
63. Scaling AL, Prossnitz ER, Hathaway HJ. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm Cancer 2014;5:146-60.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zach Lo, Yedidia-Aryeh L, Goldberg M. Estrogen and DNA damage modulate mRNA levels of genes involved in homologous recombination repair in estrogen-deprived cells . J Transl Genet Genom 2022;6:266-80. http://dx.doi.org/10.20517/jtgg.2021.58
AMA Style
Zach Lo, Yedidia-Aryeh L, Goldberg M. Estrogen and DNA damage modulate mRNA levels of genes involved in homologous recombination repair in estrogen-deprived cells . Journal of Translational Genetics and Genomics. 2022; 6(2): 266-80. http://dx.doi.org/10.20517/jtgg.2021.58
Chicago/Turabian Style
Zach, Li-or, Lia Yedidia-Aryeh, Michal Goldberg. 2022. "Estrogen and DNA damage modulate mRNA levels of genes involved in homologous recombination repair in estrogen-deprived cells " Journal of Translational Genetics and Genomics. 6, no.2: 266-80. http://dx.doi.org/10.20517/jtgg.2021.58
ACS Style
Zach, L.o.; Yedidia-Aryeh L.; Goldberg M. Estrogen and DNA damage modulate mRNA levels of genes involved in homologous recombination repair in estrogen-deprived cells . J. Transl. Genet. Genom. 2022, 6, 266-80. http://dx.doi.org/10.20517/jtgg.2021.58
About This Article
Copyright

Data & Comments
Data


 Cite This Article 22 clicks
Cite This Article 22 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.