
The somatic clinico-genomics of localized, non-indolent prostate cancer


Abstract
Prostate cancer is the second most frequently diagnosed non-skin male malignancy worldwide, with over 1.1 million new diagnoses each year. Population screening based on serum prostate-specific antigen (PSA) has shifted the disease burden toward earlier stage, localized disease. However, clinical outcomes for localized prostate cancer are highly heterogeneous, despite the use of clinical prognostic factors (PSA, Gleason grade, and TNM stage). Recent advances in massively-parallel DNA sequencing and computational biology have permitted detailed genomic analyses of all major human cancer types, including prostate cancer. However, the long natural history of prostate cancer has largely precluded rapid translation of this knowledge into clinical practice. Herein, we provide a “state of the field” overview of prostate cancer genomics, with particular focus on localized, non-indolent disease. We discuss recurrent somatic aberrations, across multiple mutational classes, which characterize this disease state, and suggest strategies through which an improved understanding of these molecular aberrations may be utilized in the curative setting. Finally, we summarize the major outstanding questions in prostate cancer genomics, and discuss hypotheses and potential strategies to begin to address these questions.
Keywords
Introduction
Prostate cancer is the most frequently diagnosed non-skin male malignancy in Western countries, with over 500,000 new diagnoses each year in North America and Europe alone. Since the introduction of prostate-specific antigen (PSA) screening in the early 1990s, the vast majority of new cases are diagnosed as organ-confined, potentially-curable disease, with very few diagnoses of primary metastatic disease in screened populations[1]. Localized prostate cancer is triaged into groups that largely define treatment options, risk of disease progression and prostate cancer-specific mortality (PCSM). These risk groups (i.e., low, intermediate, high) are based primarily on three clinical prognostic factors: serum PSA concentration, Gleason/International Society for Urological Pathology grade of the tumour biopsy, and clinical TNM stage[2]. Low risk disease can usually be effectively managed through active surveillance. Intermediate risk prostate cancer is generally treated with curative-intent radical prostatectomy or image-guided radiotherapy (IGRT; external-beam and/or high dose-rate brachytherapy), while high risk disease is managed with IGRT and (neo)adjuvant androgen-deprivation therapy (ADT) or radical prostatectomy, with or without adjuvant IGRT[3].
Despite these prognostic factors, clinical outcomes are highly heterogeneous across the risk spectrum. For example, 20%-25% of men on active surveillance for low risk prostate cancer will experience clinical or pathological progression, necessitating definitive therapy[4]. Similarly, 10%-15% and 30%-40% of men with intermediate or high-risk prostate cancer, respectively, will experience disease recurrence within 3 years following curative-intent therapy, portending a lethal clinical course[5]. As such, current prognostic factors do not accurately define the risk of disease progression for an individual man with prostate cancer. There is, therefore, an urgent need to define novel prognostic and predictive biomarkers to inform precision medicine protocols for localized prostate cancer. Such biomarkers would allow for more appropriate pre-treatment triage, thus reducing over-treatment of indolent prostate cancer and intensifying treatment for men at high risk for progression to potentially-lethal metastatic disease.
The prostate gland
The prostate is an androgen-dependent tissue, involved in normal reproductive function. Chemical or physical castration of mice results in complete regression of the prostate, which is rescued by exogenous androgen[6]. Testosterone is primarily synthesized from cholesterol in the Leydig cells of the testis (a small amount is synthesized in the adrenal gland), and is converted to 5-alpha-dihydrotestosterone (DHT) in androgen-responsive tissues such as the prostate. DHT has a 2-5-fold higher affinity for the androgen receptor (AR) and a 10-fold higher effect on AR-induced gene expression than testosterone. AR is a prototypical steroid hormone receptor, which, when bound by androgen, translocates into the nucleus and interacts with androgen-responsive elements (ARE) in the promoter and enhancer regions of target genes such as TMPRSS2 and KLK3, which promote proliferation and survival of prostate epithelial cells[7].
Genomic drivers of prostate cancer tumorigenesis and progression
An understanding of the molecular features of prostate cancer requires dissection of the differential biology of primary, treatment-naïve, potentially-curable localized disease vs. incurable metastatic, castration-resistant prostate cancer (mCRPC) that has evolved in the context of multiple rounds of ADT. Primary prostate cancer is a C-class tumor[8], characterized by a paucity of driver single-nucleotide variants (SNVs) with recurrent driver structural variants, including DNA copy number aberrations (CNAs) and genomic rearrangements (GRs)[9-11]. Conversely, mCRPC is associated with increased genomic instability and mutational burden, and with enrichment of gene mutations which drive androgen-independent growth and metastatic dissemination[12-14]. This review will focus upon genomic aberrations associated with localized, potentially-curable disease, with a particular emphasis on molecular aberrations that define novel disease subtypes and clinically-relevant biomarkers of aggressive localized disease. While epigenomic aberrations are highly relevant driver events in localized prostate cancer and may have substantial value within prognostic signatures, a detailed discussion of prostate cancer epigenomics is beyond the scope of this review, which will focus predominantly on genomic aberrations that result in changes in DNA sequence or structure. Similarly, cancer evolves in the context of unique germline genotypes, which can fundamentally alter tumour somatic phenotypes and aggression; a classic example is the unique somatic molecular profile of aggressive prostate cancer in men harbouring a germline mutation in the BRCA2 gene[15]. While this review will focus on somatic aberrations, the influence of germline background is highly relevant, as evidenced by the preponderance of genomic loci linked to the onset and aggression of localized prostate cancer[16-21].
The molecular landscape of prostate cancer
GRs
The origins of our understanding of the molecular determinants of prostate cancer tumorigenesis and aggression predates the current genomic era. The effectiveness of androgen ablation in the treatment of prostate cancer has been recognized for decades[22], and this association strongly (and correctly) suggested a link between AR activity and prostate cancer progression[23]. Clinical studies revealed that AR mutations are common in advanced prostate cancer and arise during the course of ADT[24,25].
A major breakthrough came with the discovery that the ETS-family oncogene, ERG, is over-expressed in a large proportion of primary prostate cancers[26], and the subsequent finding that this is secondary to an androgen-dependent GR on chromosome 21q22.2-22.3 that results in a fusion between the 5’ regulatory region of the TMPRSS2 gene and the coding region of ERG[27][Figure 1]. This fusion results from one of two independent-but-related processes: homozygous deletion of the intervening ~2.8 Mbp between the TMPRSS2 and ERG genes (termed “edel”) or translocation of the intervening region to other chromosomal locations (termed “esplit”)[28]. These can be distinguished using three-colour fluorescence in situ hybridization (FISH) analysis, and edel fusions can also be inferred from copy number loss of the intervening genomic region (see below).
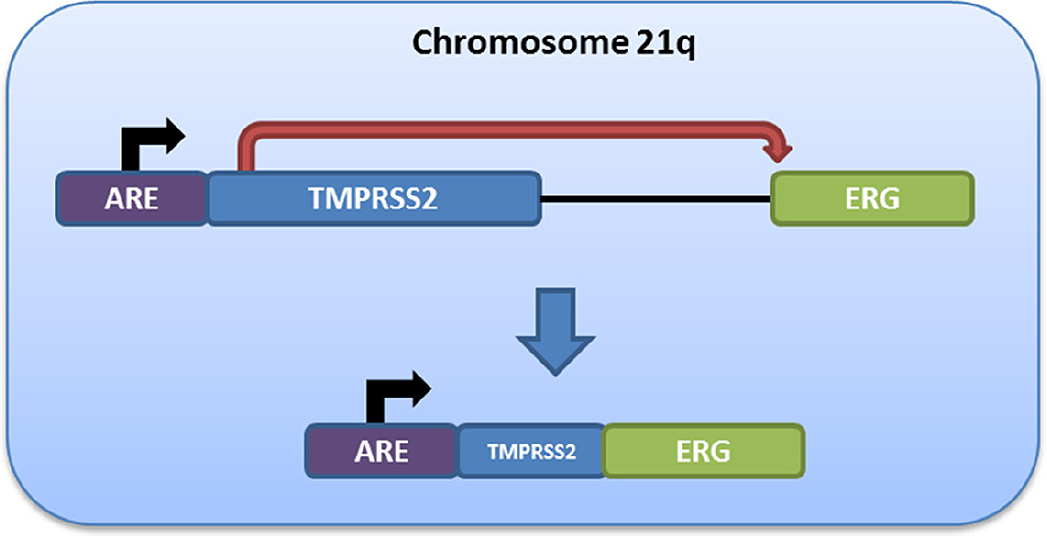
Figure 1. Schematic of androgen-driven ERG expression following TMPRSS2:ERG fusion. Fusion of the 5’ regulatory region of the TMPRSS2 gene (21q22.3) - including an upstream androgen response element (ARE) - to the coding region of the ERG oncogene (21q22.2) results in a fusion product in which ERG expression is induced in an androgen-responsive manner. This fusion can be detected using fluorescence in situ hybridization[28] or indirectly through ERG immunohistochemistry
The TMPRSS2:ERG fusion (T2E) is present in ~45% of all localized prostate cancers, while another ~5%-10% harbor a fusion involving other ETS-family proto-oncogenes (e.g., ETV1, ETV4)[29]. Fusion of the TMPRSS2 promoter, which contains a strong ARE, to the 5’ end of ERG leads to androgen-driven ERG overexpression, which can be detected in clinical specimens by immunohistochemistry against ERG[30,31]. The T2E fusion is also readily detectable using FISH and qPCR[28,32]. Molecular heterogeneity and subclonality studies have consistently demonstrated that the T2E fusion is one of the earliest molecular events in prostate tumorigenesis[33]. Despite this, no clear picture has emerged regarding the precise function of T2E in this regard. Moreover, large studies have failed to establish a prognostic effect of T2E for differential clinical outcomes in localized disease[34,35], although tumors harboring the fusion do show unique transcriptional programming resulting in a dependency on NOTCH signaling[36], which could make T2E a predictive biomarker for sensitivity to NOTCH pathway inhibitors.
Similarly, recent evidence indirectly links T2E to resistance to ADT. In mCRPC treated with the second-line ADT agent abiraterone, patients whose disease harbored SNVs in the SPOP gene or deletions of CHD1 (see below) had significantly prolonged overall survival times, relative to patients without either aberration[37]. SPOP SNVs/CHD1 deletions were mutually exclusive from T2E fusions, as previously described[38,39], suggesting that T2E may predict resistance to abiraterone-based ADT. Further studies of T2E in this context will be required to determine whether fusion-negative tumors have differential abiraterone sensitivity in the context of SPOP/CHD1 aberrations.
While ETS-family fusions represent the largest single class of prostate cancer-associated GRs, other aberrations have been described and may impact prostate cancer biology and clinical outcome. For example, a recurrent genomic inversion on chromosome 7q23 containing the PTEN tumor suppressor gene is associated with a significant reduction in PTEN mRNA abundance and PTEN function, similar to that observed in tumors harboring PTEN deletions[10]. A similar effect was also observed on a region of chromosome 3, suggesting that this is a relatively common mechanism of mRNA abundance regulation. PTEN displays the behavior of a haploinsufficient tumor suppressor, whereby inactivation of a single PTEN allele by deletion (see below) or point mutation is sufficient to drive PTEN-mediated tumourigenesis[40]. However, this effect may, in fact, be explained by a copy-neutral loss of the second allele through methylation-induced silencing or genomic inversion. As the number of tumors profiled continues to increase (thus increasing statistical power), it will be important to evaluate if and how different classes of aberration interact to dysregulate PTEN.
Additional non-ETS gene fusions have been identified in prostate cancers in non-Caucasian populations, including fusions between the USP9Y and TTTY15 genes on chromosome Y and a CTAGE5:KHDRBS3 fusion resulting from a chr14:chr8 translocation[41]. In Chinese prostate cancer patients, these fusions are present at rates that exceed that of TMPRSS2:ERG, which strongly suggests that ethnicity and environmental factors play a key role in the molecular progression of prostate tumorigenesis.
It is now clear that GRs, including (but not limited to) ETS family fusions, are key drivers of prostate cancer development and progression. However, if and how rearrangements contribute to patient-specific clinical aggression remains unclear.
CNAs
Several genomic loci are recurrently amplified or deleted in localized prostate cancer [Table 1]. Among the most frequent CNA is the deletion of a locus on chromosome 10q containing the PTEN tumour suppressor[9,10,29]. This CNA is present in roughly 20% of all localized cancers and is distributed equally between tumour phylogenetic trunks and branches[9]. PTEN-/- mouse embryonic fibroblasts are sensitive to poly-ADP ribose polymerase (PARP) inhibitors[42-45], possibly through decreased homologous recombination-mediated DNA repair via transcriptional down-regulation of RAD51[45,46]. However, correlative studies have failed to establish any link between PTEN status and RAD51 expression in prostate cancer[47], and it is likely that this effect is tissue-specific[43-45], since follow-up clustered regularly interspaced short palindromic repeats (CRISPR)-based screens for PARP inhibitor sensitivity have failed to recapitulate the importance of PTEN in this regard[48]. Other recurrent somatic deletions include NKX3-1 (8p21.2), CHD1 (5q15-q21.1; see below), CDH1 (16q22.1), RB1 (13q14.2), CDKN1B (12p13.1), BRCA2 (13q13.1) and TP53 (17p13.1)[9,10,29], and several of these may have significant prognostic value for adverse outcomes in localized disease[49].
Recurrent copy number aberrations in localized prostate cancer
| Gene | Aberration | Chromosomal locus | Frequency in localized prostate cancer[10,29,87] |
|---|---|---|---|
| PTEN | Deletion | 10q23.31 | 10%-20% |
| FOXO1 | Deletion | 13q14.11 | 5%-15% |
| RB1 | Deletion | 13q14.2 | 5%-15% |
| CHD1 | Deletion | 5q15-q21.1 | 8%-10% |
| MYC | Amplification | 8q24.21 | 6%-10% |
| NBN | Amplification | 8q21.3 | 5%-7% |
| TP53 | Deletion | 17p13.1 | 4%-7% |
| CDH1 | Deletion | 16q22.1 | 4%-5% |
| NKX3-1 | Deletion | 8p21.2 | 2%-5% |
| BRCA2 | Deletion | 13q13.1 | 3%-5% |
| CDKN1B | Deletion | 12p13.1 | 2%-5% |
| BRCA1 | Deletion | 17q21.31 | 1%-2% |
The most frequently amplified region of the somatic prostate cancer genome is chromosome 8q. Several significant amplification peaks exist on this chromosomal arm, the most prominent of which (8q24.21) harbours the MYC oncogene. This region is amplified in 8%-10% of localized cancers[9,10,29], and is prognostic of adverse clinical outcomes following definitive therapy[10,50-52]. Interestingly, the rate of MYC amplification is substantially increased in localized prostate cancers in men who harbour a deleterious germline mutation in the BRCA2 gene[15,53]. Consistent with a role for MYC amplification as a determinant of poor clinical outcome, these familial cancers have an extremely aggressive clinical course, with 5-year overall survival rates approaching 50%[54].
Recurrent amplifications of chromosome 8q - outside of those affecting the MYC gene[29] - are also commonly observed and may have important biological and clinical relevance[29]. For example, amplification of the NBN gene (8q22.1), which encodes the DNA damage sensor protein NBS1, is predictive of poor response to external beam radiotherapy in low/intermediate risk prostate cancer[55]. Similarly, the PCAT1 long non-coding RNA gene, which is implicated in aggressive localized and metastatic prostate cancer, is located on a frequently-amplified region of chromosome 8q immediately upstream of the MYC locus[56]. Intriguingly, PCAT1 lies within a common fragile site (FRA8C)[57], which may help to explain the recurrent instability at this locus.
Beyond chromosome 8q, recurrent amplifications have been identified on chromosome 3q26, 11q13, and the entirety of chromosome 7[10,29]. Because these amplifications involve megabase stretches of the genome, identification of the putative driver genes in each region has proven difficult. However, it is clear that at least some of these large-scale amplifications have prognostic value in localized prostate cancer; indeed, amplification of chromosome 7 defines a unique CNA-based cluster of prostate cancer, and is associated with decreased time to biochemical relapse in men treated with IGRT for intermediate risk prostate cancer[58].
The prognostic importance of somatic CNAs is now well-established. CNA burden - the proportion of the genome affected by a CNA - is associated with reduced time to biochemical relapse in men with localized prostate cancer[58,59]. Moreover, unbiased, machine learning-based approaches have identified CNA-based gene signatures that accurately classify patients for risk of biochemical and metastatic relapse following definitive, curative-intent therapy for localized prostate cancer[10,58,60]. These signatures significantly outperform RNA-based classifiers[58] as well as established clinical prognostic factors such as Gleason grade and pre-treatment PSA. Moreover, CNA-based classifiers can be derived from pre-treatment biopsy specimens and are readily adapted for clinical implementation via clinical laboratory improvement amendments-compatible platforms such as NanoString[60].
Single nucleotide variants
In contrast to other solid tumour types such as serous ovarian and pancreatic cancers, localized prostate cancer harbours a paucity of recurrent driver somatic SNVs. Indeed, several well-powered studies have identified very few genes mutated at rates exceeding 5%. The most frequently mutated gene in localized prostate cancer in SPOP, which encodes a transcriptional regulator implicated in the DNA damage response, maintenance of genome integrity, and inactivation of signaling pathways involved in cell proliferation and survival[10,38,61,62]. SPOP mutation frequently co-occurs with CHD1 deletion and both events are mutually exclusive from TMPRSS2:ERG fusion. While the understanding of the molecular mechanism for these associations is incomplete, recent data suggest that impairment of androgen signaling in cells lacking CHD1 or SPOP is likely a major contributor. CHD1 is required for androgen-dependent TMPRSS2:ERG fusion[63] and SPOP inhibits androgen signaling via ubiquitylation of the AR steroid receptor coactivator-3 and of AR itself[64]. Recent evidence suggests that SPOP mutation is subject to negative selection during the development of mCRPC, perhaps reflecting the reduced dependence on androgen signaling in this disease state. From a therapeutic standpoint, SPOP mutation (and CHD1 deletion) are predictive of improved response to the anti-androgen abiraterone[37].
Mutations in TP53 are also relatively common in localized prostate cancer (3%-5% recurrence), and these tend to cluster in the central DNA binding domain of p53[10], suggesting an important functional contribution of this canonical tumour suppressor pathway. Similarly, mutations in the ATM gene, while somewhat rarer than those in TP53 (~2% recurrence), are strongly associated with rapid biochemical relapse in localized disease[10]. The mechanisms underlying this clinical aggression are unclear, since the majority of these mutations do not map to established hotspots or functional domains within the ATM protein. Nevertheless, the finding that ATM is frequently mutated in the germline in patients with mCRPC[65,66] underlies the importance of this pathway as a determinant of prostate tumourigenesis and progression. Moreover, ATM mutant cancers may be uniquely sensitive to PARP inhibitors[67], suggesting a potential role as a predictive biomarker for these mutations.
Other genes affected by recurrent SNVs include MED12[38], which is implicated in the clinical aggression of hereditary prostate cancer in men who carry a germline BRCA2 mutation[15], and FOXA1, which encodes a Forkhead family transcription factor and is associated with poor outcomes in ER+ breast cancer[68].
One intriguing category of SNVs are those that occur in non-coding regions of the genome (ncSNVs). Several ncSNVs are recurrent at rates that approach those of non-synonymous SNVs (i.e., 2%-4%)[10]. These likely represent true hotspot mutations because they occur recurrently at the same nucleotide. While the precise role of these ncSNVs remains unclear, it is possible that they interfere with transcriptional regulation, either through cis-mediated effects on nearby genes or via alterations in three-dimensional genome structure. CRISPR-based knock-in models will greatly enhance our understanding of the biology of these recurrent aberrations.
The whole-genome landscape of prostate cancer
The first deep analysis of the prostate cancer genome [Table 2] emerged in 2011. Garraway and colleagues sequenced the whole genomes of seven localized prostate cancers (and patient-matched normal specimens) to a mean depth of ~30x, sufficient to detect most clonal mutations[69]. This group demonstrated that localized prostate cancer harbours an intermediate SNV burden, relative to other solid tumour types. While this study lacked sufficient statistical power to detect all but the most recurrent mutations, several genes, including SPTA1 and SPOP, were altered above the expected background rate. This report also identified a unique pattern of GR in prostate cancers in which closed loops of rearrangements occur in a copy-neutral manner to generate complex fusion products that dysregulate numerous genes, including cancer-associated genes such as TP53, ABL1, and TBK1. These rearrangements were enriched in ChIP-seq peaks associated with open chromatin, active transcription and both AR and ERG binding. GRs were also identified in other prostate cancer-associated genes, such as PTEN[70].
Summary of whole genome sequencing studies of localized prostate cancer
| Ref. | Number and type of whole genomes sequenced | Major significance |
|---|---|---|
| Berger et al.[69], 2011 | 7 primary high risk tumours | First whole-genome sequencing of localized prostate cancer. Identification of closed-loop chain rearrangements |
| Baca et al.[71], 2012 | 55 primary tumours, 2 neuroendocrine metastases | Characterization of temporal changes in prostate cancer structural variation (“chromoplexy”) |
| Weischenfeldt et al.[72], 2013 | 11 early-onset primary tumours 7 elderly-onset primary tumours | Androgen-dependent structural variation enriched in prostate cancers arising in men < 50 years of age |
| Cancer Genome Atlas Research Network[29], 2015 | 19 primary tumours | Molecular subclasses of localized prostate cancers |
| Boutros et al.[49], 2015 | 23 malignant foci from 5 primary tumours | Spatial heterogeneity of localized prostate cancer |
| Cooper et al.[77], 2015 | 12 malignant foci from 3 primary tumours | Spatial-temporal heterogeneity of localized prostate cancer. Identification of aberrations in morphologically-normal prostate epithelium |
| Fraser et al.[10], 2017 | 200 intermediate risk primary tumours | Largest study of prostate cancer whole genomes to date. Identification of recurrent driver aberrations linked to adverse clinical outcome |
| Taylor et al.[15], 2017 | 19 disease foci from 14 germline BRCA2 mutation carriers | Tumour genomes of BRCA2 mutation carriers closely resemble those of castration-resistant metastatic disease. MED12/MED12L pathway as driver of clinical aggression |
| Camacho et al.[88], 2017 | 103 primary tumours | Assessment of somatic genome-wide copy number aberrations and mechanism of copy number loss |
| Ren et al.[89], 2018 | 65 primary tumours from Chinese men | Low frequency of TMPRSS2:ERG fusion in Chinese prostate cancers. Identification of novel tumour suppressor genes |
| Espiritu et al.[9], 2018 | 93 intermediate risk primary tumours | Analysis of the temporal evolution of prostate cancer. Development of a clonality-aware multi-modal biomarker of adverse clinical outcome |
| Wedge et al.[79], 2018 | 87 primary tumours, 20 metastatic lesions | Temporal evolution of prostate cancer. Identification of potential druggable targets in localized disease |
| Su et al.[84], 2018 | 17 nuclei from 2 primary tumours | First report of single nucleus whole-genome sequencing in prostate cancer. Significant spatial heterogeneity within the same gland |
A subsequent study from the same group[71] showed that these complex closed-loop chain rearrangements occur through a process termed “chromoplexy”. Using simulated genome data, the authors demonstrated that chromoplexy is an important mechanism of tumour suppressor gene inactivation in prostate cancer, and further demonstrated that chromoplexy occurs throughout the subclonal evolution of prostate cancer in a “punctuated equilibrium” of tumour evolution.
The Cancer Genome Atlas (TCGA) program sequenced the whole exomes of 333 primary prostate cancers, with whole-genome sequencing of 19 cases of low mutational burden[29], representing the first well-powered survey of protein coding variants in localized prostate cancer. Using associated CNA, RNA expression, DNA methylation, and protein phosphorylation arrays, the TCGA team identified several consensus clusters of localized prostate cancer, including tumours harbouring an ETS fusion (~60%) and SPOP, FOXA1, or IDH1 mutation (~15%, collectively). Approximately 25% of localized prostate cancers did not sort into one of these clusters, perhaps suggesting the existence of additional molecular subtypes that could not be detected given the statistical power of the TCGA study and the relatively low mutational burden in localized prostate cancer. While TCGA identified molecular aberrations in several clinically-relevant pathways (e.g., PI3K, DNA repair, and other), the putative effects of these aberrations on clinical outcome could not be assessed due to the limited clinical follow up associated with the genomic findings. One potential clinical utility of Weischenfeldt et al.[72] surveyed the whole genomes of eleven prostate cancers arising in men 50 years of age or younger and compared these with seven propensity-matched prostate cancers of elderly-onset. While rare, these cancers in younger men represent a unique clinical challenge, and both the long natural life expectancy of these men and the increased clinical aggression of these early-onset cancers necessitates definitive local therapy. Interestingly, these cancers harbour a lower overall burden of structural variation, perhaps owing to the reduced time for acquisition of mutations, but possess a strong bias toward androgen-induced GRs - such as T2E - relative to cancers arising in older men. This was associated with increased AR mRNA abundance in early-onset prostate cancer. These findings suggest a unique biology of early-onset prostate cancer and pave the way for future studies to define the precise molecular mechanisms through which these cancers develop and evolve.
Clinico-genomics in localized prostate cancer
Well-powered, clinically-focused studies of the localized prostate cancer genome have proven difficult, relative to other solid tumour types, because of both the paucity of high tumour content tissue, particularly in low burden disease. Moreover, the development of clinically-meaningful endpoints (i.e., distant metastasis-free survival, overall survival, etc.) in localized prostate cancer can take upward of 10-15 years. However, recent advances in computational algorithms and decreases in sequencing costs have allowed for far more in-depth analyses than have previously been possible. Moreover, recent work has established reliable surrogate endpoints of aggressive disease.
Fraser et al.[10] performed a deep meta-analysis of prostate cancer whole-genome (n = 200) and whole-exome (n = 477) sequences published to date, with long-term clinical follow-up available for 130 patients with homogeneously staged disease. Using parallel array-based indices, the team identified a 6-feature clinico-genomic signature of adverse clinical outcome[10]. This signature is composed of a GR (a translocation on chromosome 7), two DNA methylation aberrations (TCERG1L hypomethylation, ACTL6B hypermethylation), SNVs in ATM, copy number amplification of MYC, as well as clinical T stage. This signature accurately predicts overall biochemical relapse and, importantly, also predicts relapse within the first 3 years following curative-intent treatment, which is a surrogate for lethal disease[5,73]. Despite these promising insights, much larger numbers of patients - with well-annotated long-term clinical follow-up data - will be required to both validate these findings and to establish the clinical implications of aberrations occurring below the 1%-5% recurrence threshold.
Spatio-temporal heterogeneity, tumour evolution, and clinical outcomes approximately 75%-80% of prostate cancers are multi-focal, as assessed at prostatectomy[74,75]. While multiple foci do not necessarily portend a more aggressive clinical course[74], the index lesion does not always correspond to the malignant clone that gives rise to distant metastases[76]. As such, an understanding of if and how inter-focal heterogeneity affects prostate cancer progression is of paramount importance. To that end, Boutros et al.[49] sequenced the genomes of eighteen spatially-distinct disease foci from five patients with intermediate-risk prostate cancer, with an additional sixty-nine cases evaluated using array-based assessment of genome-wide CNAs. Inter-focal heterogeneity was extremely high for all mutational classes. For example, in one patient, the total number of CNAs varied by nearly 2 orders of magnitude across five tumour foci, while the percentage of the genome affected by CNAs (percentage of genome altered; a metric of genomic instability and an independent prognostic factor[58,59]) varied by over 10-fold. Importantly, the index lesion was not universally associated with the highest burden of structural variants or SNVs, consistent with reports showing that metastases do not always arise from the index tumour[76]. Moreover, several aberrations with established links to progression and adverse outcomes (e.g., TP53 and PTEN deletion) were heterogeneous across tumour foci from the same patient, and one patient showed strong evidence of multiple independent tumours. Likewise, Cooper et al.[77] sequenced the whole genomes of twelve cancer foci from three patients, along with tumour-adjacent normal prostate epithelium from all three. The team showed compelling evidence of intermixing of cancer clones within a single cancer focus and identified a significant burden of tumour-associated mutation in morphologically normal prostate epithelium, consistent with a global “field defect” within the gland as a whole. These findings of extensive heterogeneity have subsequently been recapitulated in larger, independent cohorts. Løvf et al.[78] recently sequenced the exomes of 153 foci from 41 patients, including both malignant and putatively-normal prostate epithelium, and demonstrated that there is significant inter-focal heterogeneity, including genes with established prognostic importance or enrichment in metastatic or aggressive localized prostate cancers (e.g., NKX3-1, MED12, TP53).
Prostate cancer also shows evidence of substantial clonal divergence during tumourigenesis. Indeed, Espiritu et al.[9] recently demonstrated that only ~25% of localized, non-indolent prostate cancers are composed of a single clonal population (“monoclonal”) within the index lesion. Conversely, 75% of cancers showed strong evidence of two or more clonal populations (“polyclonal”), which diverged into unique clonal “branches” early in the process of tumourigenesis and share very few driver aberrations (shared “trunk” mutations). Interestingly, patients who harbor monoclonal tumours almost never experienced disease relapse. Moreover, polyclonal tumours were more aggressive, and this aggression was synergistic with established molecular predictors of adverse outcome, such as the Fraser 6-feature signature (see above) and percentage genome alteration. These data strongly support the hypothesis that clonal evolution is a key determinant of clinical aggression.
Wedge et al.[79] similarly identified extensive subclonal heterogeneity and evolutionary divergence in both localized and metastatic prostate cancer. In particular, this group found that ETS-fusion status was a significant predictor of somatic CNAs at several loci, including amplification of 8q and deletion of the PTEN locus on chromosome 10. Conversely, tumours lacking a truncal ETS fusion were enriched for CHD1 deletion - consistent with the previous finding that T2E and CHD1 deletion are mutually exclusive - as well as CNAs at several driver loci, including BRCA2, RB1, and FOXO1. Intriguingly, subclonal analyses revealed that metastatic tumours are significantly less molecularly heterogeneous than primary tumours, consistent with selection of a small number of aggressive subclones during metastatic spread.
These findings of subclonal divergence have important potential consequences for the clinical use of molecular biomarkers. While no study has comprehensively evaluated the rates of metastatic seeding from the index lesion vs. independent foci, there is evidence that the highest-grade focus does not always seed the “lethal clone”[76]. As such, clinical biomarkers based on single disease foci - such as would be obtained from an index lesion-derived biopsy - may not accurately reflect the aggressive potential of the entire prostate. Despite this, biomarkers based on single tumour foci have shown excellent prognostic accuracy (AUCs of 0.85 or higher). While this may reflect a deficiency in the biomarker itself, it is also possible that biomarker accuracy is capped at ~85% due to inadequate profiling of multiple tumour foci. This is a major outstanding question in translational cancer genomics, and a robust understanding of the effects of heterogeneity will be required to optimize any tissue-based biomarker that ultimately achieves clinical implementation. One potential solution is the use of liquid-based biomarkers to complement tissue biomarkers, since these may better reflect the global mutational profile across tumour foci.
Toward clinical implementation of somatic tumour genomics in prostate cancer localized prostate cancer is associated with a very low level of recurrent non-synonymous mutation. Prostate cancer lacks the type of near-ubiquitous driver aberrations - frequently seen in other cancers such as chronic myeloid leukemia (BCR-ABL fusion), pancreatic cancer (KRAS mutation), or serous ovarian cancer (TP53 mutation) - which would suggest potential novel therapeutic targets. TMPRSS2:ERG fusion is present in ~40% of all localized prostate cancers, and peptidomimetic inhibitors of ERG have been developed[80], although none have yet moved beyond the pre-clinical stage and the potential for deleterious effect of ERG inhibition on non-prostate tissues may limit their therapeutic potential.
Moreover, traditional hypotheses regarding the utility of cancer genomics to identify potential drug targets may not be as relevant to localized prostate cancer as in other cancer types or, indeed, in metastatic prostate cancer. As noted, in PSA-screened populations, virtually all new cases of prostate cancer are diagnosed as localized disease, with no evidence of extra-prostatic spread by bone scintigraphy and are thus potentially curable. While 10-year prostate cancer-specific survival for localized disease approaches 99% irrespective of treatment modality[81], biochemical and metastatic relapse-free rates are more variable. These endpoints are established surrogates of PCSM[5,73], and the heterogeneity of outcome based on these endpoints has been borne out in longer-term studies[82]. Moreover, these endpoints represent points of clinical intervention, which can result in significant, life-long morbidities secondary to salvage therapy and chemical castration. Thus, given the curative potential of local therapy and the significant adverse effects of long-term salvage therapy after treatment failure, it is imperative that techniques be developed to better classify risks of treatment relapse in the localized setting. To that end, the potential utility of intrinsic tumour genomics as predictive or prognostic biomarkers has been explored by several groups.
CNA burden is an established independent prognostic factor for biochemical relapse[58,59], although recent evidence suggests that multi-omic profiling may more accurately classify aggressive disease than assessment of individual analytes alone[83]. To that end, a novel clinico-genomic signature, based on a CNA (MYC gain), two aberrantly methylated genomic loci (TCERG1L and ACTL6B), an SNV (ATM), an inter-chromosomal translocation (chr7:61Mbp), and clinical T stage, dramatically outperformed CNA burden for predicting rapid biochemical relapse in intermediate-risk disease[10]. Moreover, the performance of this signature is further enhanced by inclusion of tumour subclonality as a prognostic factor[9]. These signatures compare favourably to existing, FDA-approved classifiers based on mRNA abundance[58], and can be called based on tissue available in routinely-available, pre-treatment needle core biopsies[9,10,49,58,60].
Thus, while prospective validation of these biomarkers is required, it appears highly likely that genomic classifiers will vastly outperform clinical prognostic factors and have the potential to revolutionize treatment stratification for localized prostate cancer. Most prostate cancers are multi-focal[49,77], and there is substantial genomic heterogeneity associated with separate disease foci[49,77,79,84], including in genes with established prognostic value (e.g., MYC gain)[49]. Nevertheless, genomic signatures derived from the largest (index) lesion can predict disease aggression with accuracy of at least 85%[10], far exceeding the performance of established clinical prognostic factors. As such, whatever the influence of spatial heterogeneity on prognosis, it is largely accounted for within a single disease focus, although there are clearly cases where this relationship does not hold[76]. Thus, the question arises whether enhancements to prognostic signatures based on intrinsic genomics of single loci will further improve biomarker performance- due to inclusion of novel analytes, deeper profiling of rare variants, or both. It is likely that for a subset of prostate cancers, clinical aggression cannot be accurately predicted from the index lesion[76], and thus profiling of additional foci will be required for accurate triage. One possible solution to this problem is a movement toward the so-called liquid biopsy, which would reflect the aggregate of all cancer-associated aberrations and thus would, in theory, overcome genomic heterogeneity. Indeed, extra-prostatic extension can be accurately identified based on proteomic analysis of post-digital rectal examination urine[85], supporting the viability of this approach.
Ultimately, clinical trials are required to validate the ability of these tools to improve treatment outcomes for localized prostate cancer. One potential model is to provide a “signature score” for all patients; patients whose signature score portends a favourable clinical outcome would receive standard of care therapy, while those with adverse signature scores would be randomized to receive either standard of care or intensified therapy [Figure 2]. While the specific intensification strategies to be employed are beyond the scope of this review, they may involve additional therapies that are already approved for treatment of localized prostate cancer. For example, a man with intermediate risk disease who would ordinarily be treated with IGRT alone might instead receive radiotherapy plus neoadjuvant androgen deprivation.
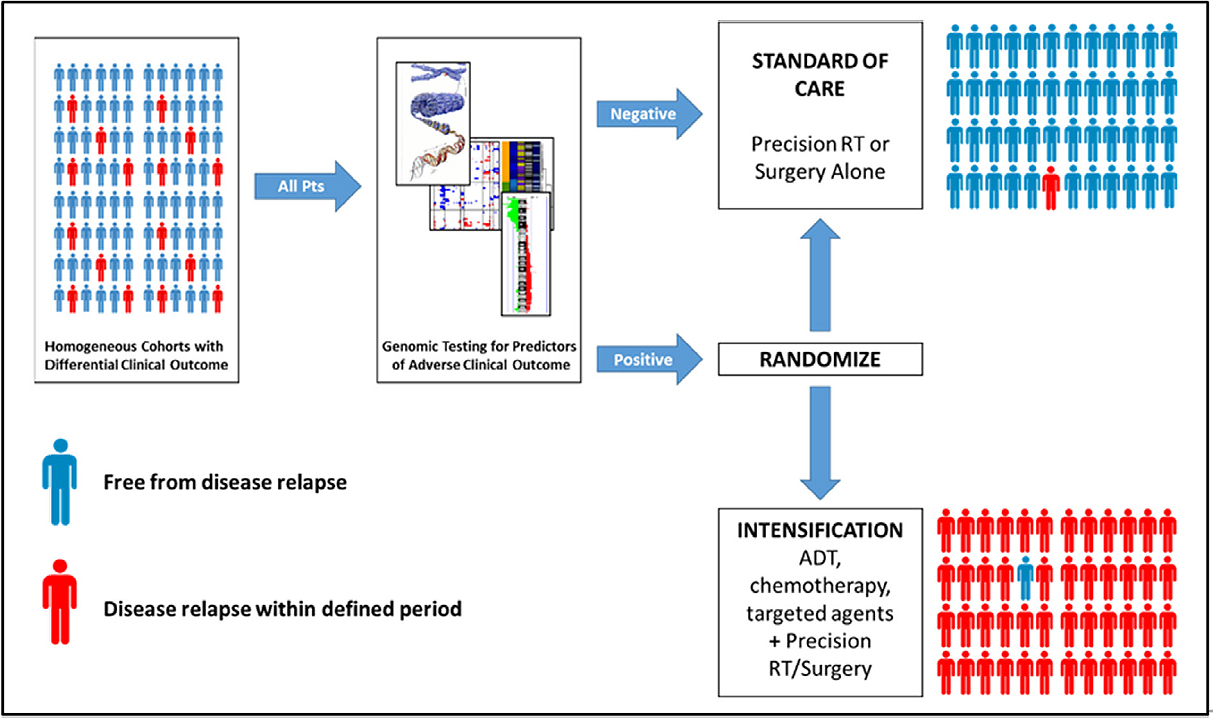
Figure 2. A model for clinical trials of prognostic biomarkers in localized, non-indolent prostate cancer. All patients newly diagnosed with localized prostate cancer receive a test to establish risks of disease relapse following local therapy, in addition to traditional stratification based on existing clinical prognostic factors. Patients whose test suggests a low likelihood of adverse clinical outcome receive standard of care therapy (i.e., precision radiotherapy or surgery). Patients whose test suggests a likelihood of disease relapse significantly higher than expected from clinical prognostic factors are randomized to standard of care or standard of care plus intensification, which may include (neo) adjuvant androgen deprivation (ADT), chemotherapy, or molecular targeted agents
Conclusion
The rapid improvement of second-generation sequencing technologies - and the concomitant reduction in sequencing prices - have dramatically improved our understanding of the molecular underpinnings of prostate cancer. Indeed, in the next 2-3 years, it is anticipated that discovery of somatic driver mutations in localized prostate cancer will saturate at the 0.5%-1% recurrence level. Unfortunately, very little of the currently-available genomic data are associated with long term clinical follow-up, thus potentially limiting the clinical applicability of these well-powered datasets and meta-analyses. As such, there is a need to develop novel patient cohorts - both retrospective and prospective - to facilitate biomarker discovery and validation, respectively.
Similarly, there is an urgent need to expand upon our current understanding of the molecular link between the primary tumour and distant metastases. In particular, it will be important to elucidate the effects of spatial heterogeneity and to identify features that accurately identify the lethal focus (and, indeed, the lethal subclone) from a prostate harbouring multi-focal disease.
While the genome-wide CNA and SNV landscape is well-established, improvements in long-read and linked-read sequencing will permit a more robust understanding of the nature of recurrent, large-scale structural variation in prostate cancer. Indeed, recent data have revealed heretofore occult structural variation of the non-coding genome that may, in part, help to explain the uniquely aggressive biology of mCRPC[86]. Application of these technologies will revolutionize our understanding of the complex structural variation that underlies prostate cancer, but which has been difficult to ascertain using traditional 2 × 150 bp paired-end sequencing.
These and other molecular tools will continue to revolutionize our understanding of the basic biology of prostate cancer. Moreover, as analysis costs continue to fall, the number of patients whose tumours can be comprehensively profiled will only continue to increase. It will, therefore, be possible to develop clinico-genomic portraits of patients across the disease spectrum, and thus to develop, validate, and implement molecular biomarkers of prostate cancer diagnosis and clinical outcome.
Declarations
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Zhou CK, Check DP, Lortet-Tieulent J, Laversanne M, Jemal A, et al. Prostate cancer incidence in 43 populations worldwide: an analysis of time trends overall and by age group. Int J Cancer 2016;138:1388-400.
2. D’Amico AV, Whittington R, Malkowicz SB, Schultz D, Blank K, et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998;280:969-74.
3. Litwin MS, Tan HJ. The diagnosis and treatment of prostate cancer: a review. JAMA 2017;317:2532-42.
4. Perlis N, Klotz L. Contemporary active surveillance: candidate selection, follow-up tools, and expected outcomes. Urol Clin North Am 2017;44:565-74.
5. Jackson WC, Suresh K, Tumati V, Allen SG, Dess RT, et al. Intermediate endpoints after postprostatectomy radiotherapy: 5-year distant metastasis to predict overall survival. Eur Urol 2018;74:413-9.
6. Ellwood-Yen K, Wongvipat J, Sawyers C. Transgenic mouse model for rapid pharmacodynamic evaluation of antiandrogens. Cancer Res 2006;66:10513-6.
7. Hsieh AC, Small EJ, Ryan CJ. Androgen-response elements in hormone-refractory prostate cancer: implications for treatment development. Lancet Oncol 2007;8:933-9.
8. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 2013;45:1127-33.
9. Espiritu SMG, Liu LY, Rubanova Y, Bhandari V, Holgersen EM, et al. The evolutionary landscape of localized prostate cancers drives clinical aggression. Cell 2018;173:1003-13.
10. Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541:359-64.
11. Cooperberg MR, Erho N, Chan JM, Feng FY, Fishbane N, et al. The diverse genomic landscape of clinically low-risk prostate cancer. Eur Urol 2018;74:444-52.
12. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215-28.
13. Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet 2018;50:645-51.
14. Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018;174:758-69.
15. Taylor RA, Fraser M, Livingstone J, Espiritu SM, Thorne H, et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat Commun 2017;8:13671.
16. Amin Al Olama A, Benlloch S, Antoniou AC, Giles GG, Severi G, et al. Risk analysis of prostate cancer in PRACTICAL, a multinational consortium, using 25 known prostate cancer susceptibility loci. Cancer Epidemiol Biomarkers Prev 2015;24:1121-9.
17. Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet 2013;45:385-91.
18. Amin Al Olama A, Kote-Jarai Z, Schumacher FR, Wiklund F, Berndt SI, et al. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum Mol Genet 2013;22:408-15.
19. Mijuskovic M, Saunders EJ, Leongamornlert DA, Wakerell S, Whitmore I, et al. Rare germline variants in DNA repair genes and the angiogenesis pathway predispose prostate cancer patients to develop metastatic disease. Br J Cancer 2018;119:96-104.
20. Dadaev T, Saunders EJ, Newcombe PJ, Anokian E, Leongamornlert DA, et al. Fine-mapping of prostate cancer susceptibility loci in a large meta-analysis identifies candidate causal variants. Nat Commun 2018;9:2256.
21. Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 2018;50:928-36.
23. van der Kwast TH, Schalken J, Ruizeveld de Winter JA, van Vroonhoven CC, Mulder E, et al. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int J Cancer 1991;48:189-93.
24. Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med 1995;332:1393-8.
25. Gaddipati JP, McLeod DG, Heidenberg HB, Sesterhenn IA, Finger MJ, et al. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res 1994;54:2861-4.
26. Petrovics G, Liu A, Shaheduzzaman S, Furusato B, Sun C, et al. Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene 2005;24:3847-52.
27. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644-8.
28. Yoshimoto M, Joshua AM, Chilton-Macneill S, Bayani J, Selvarajah S, et al. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia 2006;8:465-9.
29. Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011-25.
30. Tomlins SA, Palanisamy N, Siddiqui J, Chinnaiyan AM, Kunju LP. Antibody-based detection of ERG rearrangements in prostate core biopsies, including diagnostically challenging cases: ERG staining in prostate core biopsies. Arch Pathol Lab Med 2012;136:935-46.
31. Park K, Tomlins SA, Mudaliar KM, Chiu YL, Esgueva R, et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia 2010;12:590-8.
32. Mertz KD, Setlur SR, Dhanasekaran SM, Demichelis F, Perner S, et al. Molecular characterization of TMPRSS2-ERG gene fusion in the NCI-H660 prostate cancer cell line: a new perspective for an old model. Neoplasia 2007;9:200-6.
33. Baker SJ, Reddy EP. Understanding the temporal sequence of genetic events that lead to prostate cancer progression and metastasis. Proc Natl Acad Sci U S A 2013;110:14819-20.
34. Dal Pra A, Lalonde E, Sykes J, Warde F, Ishkanian A, et al. TMPRSS2-ERG status is not prognostic following prostate cancer radiotherapy: implications for fusion status and DSB repair. Clin Cancer Res 2013;19:5202-9.
35. Minner S, Enodien M, Sirma H, Luebke AM, Krohn A, et al. ERG status is unrelated to PSA recurrence in radically operated prostate cancer in the absence of antihormonal therapy. Clin Cancer Res 2011;17:5878-88.
36. Kron KJ, Murison A, Zhou S, Huang V, Yamaguchi TN, et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat Genet 2017;49:1336-45.
37. Boysen G, Rodrigues DN, Rescigno P, Seed G, Dolling D, et al. SPOP mutated/CHD1-deleted lethal prostate cancer and abiraterone sensitivity. Clin Cancer Res 2018;24:5585-93.
38. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012;44:685-9.
39. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012;487:239-43.
40. Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol 2003;1:E59.
41. Ren S, Peng Z, Mao JH, Yu Y, Yin C, et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res 2012;22:806-21.
42. Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 2009;1:315-22.
43. Forster MD, Dedes KJ, Sandhu S, Frentzas S, Kristeleit R, et al. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol 2011;8:302-6.
44. Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med 2010;2:53ra75.
45. McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res 2010;70:5457-64.
46. Gupta A, Yang Q, Pandita RK, Hunt CR, Xiang T, et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle 2009;8:2198-210.
47. Fraser M, Zhao H, Luoto KR, Lundin C, Coackley C, et al. PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res 2012;18:1015-27.
48. Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018;559:285-9.
49. Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet 2015;47:736-45.
50. Zafarana G, Ishkanian AS, Malloff CA, Locke JA, Sykes J, et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer 2012;118:4053-62.
51. Locke JA, Zafarana G, Ishkanian AS, Milosevic M, Thoms J, et al. NKX3.1 haploinsufficiency is prognostic for prostate cancer relapse following surgery or image-guided radiotherapy. Clin Cancer Res 2012;18:308-16.
52. Ishkanian AS, Zafarana G, Thoms J, Bristow RG. Array CGH as a potential predictor of radiocurability in intermediate risk prostate cancer. Acta Oncol 2010;49:888-94.
53. Castro E, Jugurnauth-Little S, Karlsson Q, Al-Shahrour F, Piñeiro-Yañez E, et al. High burden of copy number alterations and c-MYC amplification in prostate cancer from BRCA2 germline mutation carriers. Ann Oncol 2015;26:2293-300.
54. Risbridger GP, Taylor RA, Clouston D, Sliwinski A, Thorne H, et al. Patient-derived xenografts reveal that intraductal carcinoma of the prostate is a prominent pathology in BRCA2 mutation carriers with prostate cancer and correlates with poor prognosis. Eur Urol 2015;67:496-503.
55. Berlin A, Lalonde E, Sykes J, Zafarana G, Chu KC, et al. NBN gain is predictive for adverse outcome following image-guided radiotherapy for localized prostate cancer. Oncotarget 2014;5:11081-90.
56. Guo H, Ahmed M, Zhang F, Yao CQ, Li S, et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nat Genet 2016;48:1142-50.
57. Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, et al. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep 2013;4:420-8.
58. Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol 2014;15:1521-32.
59. Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A 2014;111:11139-44.
60. Lalonde E, Alkallas R, Chua MLK, Fraser M, Haider S, et al. Translating a prognostic DNA genomic classifier into the clinic: retrospective validation in 563 localized prostate tumors. Eur Urol 2017;72:22-31.
61. Blattner M, Liu D, Robinson BD, Huang D, Poliakov A, et al. SPOP mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell 2017;31:436-51.
62. Hjorth-Jensen K, Maya-Mendoza A, Dalgaard N, SigurÐsson JO, Bartek J, et al. SPOP promotes transcriptional expression of DNA repair and replication factors to prevent replication stress and genomic instability. Nucleic Acids Res 2018;46:9484-95.
63. Burkhardt L, Fuchs S, Krohn A, Masser S, Mader M, et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res 2013;73:2795-805.
64. Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res 2014;74:5631-43.
65. Mateo J, Boysen G, Barbieri CE, Bryant HE, Castro E, et al. DNA repair in prostate cancer: biology and clinical implications. Eur Urol 2017;71:417-25.
66. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016;375:443-53.
67. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 2015;373:1697-708.
68. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481:389-93.
69. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, et al. The genomic complexity of primary human prostate cancer. Nature 2011;470:214-20.
70. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 2010;24:1967-2000.
71. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, et al. Punctuated evolution of prostate cancer genomes. Cell 2013;153:666-77.
72. Weischenfeldt J, Simon R, Feuerbach L, Schlangen K, Weichenhan D, et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 2013;23:159-70.
73. Buyyounouski MK, Pickles T, Kestin LL, Allison R, Williams SG. Validating the interval to biochemical failure for the identification of potentially lethal prostate cancer. J Clin Oncol 2012;30:1857-63.
74. Noguchi M, Stamey TA, McNeal JE, Nolley R. Prognostic factors for multifocal prostate cancer in radical prostatectomy specimens: lack of significance of secondary cancers. J Urol 2003;170:459-63.
75. Wise AM, Stamey TA, McNeal JE, Clayton JL. Morphologic and clinical significance of multifocal prostate cancers in radical prostatectomy specimens. Urology 2002;60:264-9.
76. Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest 2013;123:4918-22.
77. Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet 2015;47:367-72.
78. Løvf M, Zhao S, Axcrona U, Johannessen B, Bakken AC, et al. Multifocal primary prostate cancer exhibits high degree of genomic heterogeneity. Eur Urol 2018; doi: 10.1016/j.eururo.2018.08.009.
79. Wedge DC, Gundem G, Mitchell T, Woodcock DJ, Martincorena I, et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet 2018;50:682-92.
80. Wang X, Qiao Y, Asangani IA, Ateeq B, Poliakov A, et al. Development of peptidomimetic inhibitors of the ERG gene fusion product in prostate cancer. Cancer Cell 2017;31:532-48.
81. Hamdy FC, Donovan JL, Lane JA, Mason M, Metcalfe C, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med 2016;375:1415-24.
82. Albertsen PC, Hanley JA, Fine J. 20-year outcomes following conservative management of clinically localized prostate cancer. JAMA 2005;293:2095-101.
83. Ross-Adams H, Lamb AD, Dunning MJ, Halim S, Lindberg J, et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: a discovery and validation cohort study. EBioMedicine 2015;2:1133-44.
84. Su F, Zhang W, Zhang D, Zhang Y, Pang C, et al. Spatial intratumor genomic heterogeneity within localized prostate cancer revealed by single-nucleus sequencing. Eur Urol 2018;74:551-9.
85. Kim Y, Ignatchenko V, Yao CQ, Kalatskaya I, Nyalwidhe JO, et al. Identification of differentially expressed proteins in direct expressed prostatic secretions of men with organ-confined versus extracapsular prostate cancer. Mol Cell Proteomics 2012;11:1870-84.
86. Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018;174:433-47.
87. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11-22.
88. Camacho N, Van Loo P, Edwards S, Kay JD Matthews L, et al. Appraising the relevance of DNA copy number loss and gain in prostate cancer using whole genome DNA sequence data. PLoS Genet 2017;13:e1007001.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Fraser M. The somatic clinico-genomics of localized, non-indolent prostate cancer. J Transl Genet Genom 2018;2:21. http://dx.doi.org/10.20517/jtgg.2018.27
AMA Style
Fraser M. The somatic clinico-genomics of localized, non-indolent prostate cancer. Journal of Translational Genetics and Genomics. 2018; 2: 21. http://dx.doi.org/10.20517/jtgg.2018.27
Chicago/Turabian Style
Fraser, Michael. 2018. "The somatic clinico-genomics of localized, non-indolent prostate cancer" Journal of Translational Genetics and Genomics. 2: 21. http://dx.doi.org/10.20517/jtgg.2018.27
ACS Style
Fraser, M. The somatic clinico-genomics of localized, non-indolent prostate cancer. J. Transl. Genet. Genom. 2018, 2, 21. http://dx.doi.org/10.20517/jtgg.2018.27
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.